| disease | Adrenogenital Syndrome |
Adrenogenital syndrome is an abnormal sexual characteristic caused by excessive adrenal androgens, leading to virilization in females and pseudopuberty in males. However, its clinical manifestations are difficult to determine in adult males. In females, this syndrome presents as pseudohermaphroditism, accompanied by thickening of the adrenal cortex. It was first documented by Creechio in 1866, but detailed descriptions of the disease were provided in 1905 by Seueira and in 1912 by Gallais, who first applied the term "Adrenogenital Syndrome." Today, the condition is understood to encompass virilization issues in both sexes.
bubble_chart Pathological Changes
Bilateral adrenal hyperplasia is the most common and is congenital. In rare cases, it is caused by a unilateral tumor (malignant or benign). Ectopic tumors (such as spermatic cord tumors) as the etiology of this disease are even rarer.
1. Hyperplasia: The size of the glands varies greatly, but they are generally very large. Each gland can weigh up to 30 grams, and in children, it may even be larger than the kidney. The cortex appears lobulated, with a brown cut surface and small nodules. The cells are from the zona reticularis of the cortex but are densely packed. Sometimes the zona glomerulosa also hyperplasias, possibly due to the stimulating effects of angiotensin.
2. Tumors: Tumor cells are densely packed and contain little lipid, resembling the normal zona reticularis. Histologically, distinguishing between adenoma and carcinoma can sometimes be difficult.
Table 1
| Age | Infant | Child | Adult |
| Lesion | Hyperplasia | Tumor | Hyperplasia or tumor |
| Male manifestation | Precocious puberty: pseudopuberty | No obvious signs | |
| Female manifestation | Female pseudohermaphroditism | Virilization | |
bubble_chart Clinical Manifestations
The zona reticularis secretes androgens, which are related to age and sex. The manifestations vary by sex and age:
1. Congenital adrenal hyperplasia with virilization: This condition is familial and hereditary, occurring in 1:5000 newborns. It can affect both sexes within the same family. The parents secrete excessive pregnanetriol under ACTH stimulation, affecting their children (Figure 1).
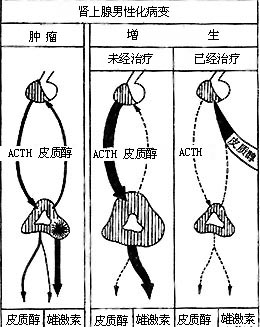
Figure 1 Pituitary-adrenal relationship in virilization
(1) In females: Early signs of virilization may be present at birth, such as clitoral hypertrophy and incomplete fusion of the urogenital folds. These girls are cases of female pseudohermaphroditism and may be mistaken for boys with hypospadias and cryptorchidism. The internal genitalia are normal but remain infantile. During development, other virilizing signs appear, such as underdeveloped breasts, absence of menstruation, and ovarian cystic changes resembling Stein-Leventhal syndrome (bilateral ovarian cystic changes).
(2) In males: No abnormalities are present at birth, but after weeks or months, the penis begins to enlarge, showing signs of isosexual precocity, though the scrotum and testes remain small. Due to adrenal androgens suppressing pituitary gonadotropins, this condition is termed macrogenitosomia praecox. Such children are described as "infant Hercules."
(3) Common changes in both sexes: Regardless of sex, skeletal development is initially abnormal. Boys initially grow rapidly, with early appearance of ossification centers and premature epiphyseal fusion, resulting in a final height rarely exceeding 150 cm. Virilization manifests as hirsutism, well-developed muscles, prominent larynx, deep voice, oily skin with acne, early penile or clitoral erections, and sexual orientation consistent with their true gender.
(1) Virilization alone: This is the most common type.
(2) Virilization with adrenal cortical insufficiency, the so-called salt-wasting type, due to 3β-hydroxysteroid dehydrogenase deficiency or absence of 11-hydroxylase.
(3) Virilization with hypertension: The rarest type, caused by 11-hydroxylase deficiency.
Adrenal cortical insufficiency: Nearly one-third of patients exhibit adrenal insufficiency. Urinary loss of Na+ and Cl-, along with virilization, may appear within the first week of life and persist for weeks. These infants are weak, expressionless, difficult to feed, and may experience vomiting, diarrhea, dehydration, and death from circulatory collapse. Acute infections may trigger adrenal crisis. Sudden death from cardiac arrest due to hyperkalemia (high K+) has been reported. This condition is more common in female infants due to noticeable external genital changes. Male infants may die before significant penile enlargement occurs, with death attributed to other causes. Diagnosis is crucial, as untreated children often die within a year.
Hypertension: A few patients develop hypertension, which can lead to cardiac enlargement and heart failure, even in young children.
3. Adrenogenital syndrome in children: If virilization occurs in male children, the cause is almost invariably a tumor. The clinical manifestations develop rapidly and, unless prenatal development was normal, they entirely resemble the congenital type. Prepubertal virilization in males is extremely rare. Testicular enlargement has also been reported, but the cause remains unclear. In very rare cases, it may be caused by adrenal cortical hyperplasia. The findings on examination are the same as in the congenital type. The pathology may be entirely similar to the congenital form.
Adrenogenital syndrome in adult females: After puberty, the original lesion continues to be active, or a new lesion appears. Cortical hyperplasia is the most common cause, occurring between 18-20 years of age, with the same etiology as the congenital type due to 21-hydroxylase deficiency. Tumors are uncommon, and if present, they occur in adulthood. The clinical manifestations vary and can be divided into the following three types:

(1) Defeminization: This results from hypothalamic and pituitary suppression. Sexual characteristics and body development are normal. Menstruation is often regular in the initial years but gradually becomes prolonged and reduced, ovulation ceases, followed by infertility and eventual menopause. In some cases, menstruation remains regular, but the primary complaint is infertility. In severe cases, breast atrophy, reduced subcutaneous fat, loss of feminine features, and even vulvar atrophy may occur, though the clitoris remains unaffected.
(2) Virilization type: This is due to the effects of androgens. Hirsutism is the most common sign, along with oily skin and acne. In severe cases, voice changes, muscle development, and clitoral enlargement may occur.
(3) Metabolic disorder type: This type is rare. Symptoms may include salt and water retention and hypertension. Insufficient corticosteroids do not cause this lesion in adults, and metabolic disorders do not manifest.
Hirsutism: Hirsutism is a significant sign of adrenogenital syndrome. In women, excessive hair growth may appear on the sides of the face, chin, and upper lip, resembling male patterns. In severe cases, women may even need to shave. On the torso, hair may grow around the nipples and along the midline of the chest and abdomen. Pubic hair distribution resembles that of males. In rare cases, excessive hair may cover the torso and limbs, similar to males. The scalp hair is thick, oily, and dark; some may experience alopecia areata at the forehead and temples, resembling older males.
Mixed type of Cushing's syndrome and this disease: This is even rarer, such as virilization accompanied by hypertension, diabetes, or what some describe as "diabetes of bearded women." Similarly, hirsutism is present in Cushing's syndrome. These mixed manifestations are particularly common in adrenocortical carcinoma.
Diagnosing this disease is not easy. Sometimes, even after thorough, meticulous, and time-consuming examinations, a conclusion cannot be reached. However, without a correct diagnosis, proper management is impossible. First, it is necessary to determine which patients require comprehensive examinations. Generally speaking, those with obvious clinical manifestations requiring treatment need thorough evaluation. Of course, examination is necessary when a tumor is suspected; on the other hand, if symptoms appear in childhood or the onset is sudden in adulthood, a tumor should be suspected. The following questions must be answered: ① Is there adrenal dysfunction? ② If so, is it hyperplasia or a tumor? ③ If it is a tumor, is it benign or malignant? On which side?
1. In females: For female pseudohermaphroditism, the diagnosis can be confirmed based on the following examinations:
(1) Microscopic examination of the cell nucleus reveals chromatin positivity. Chromosome counting shows a sex chromosome karyotype of XX.
(2) Examination of the urogenital sinus reveals the presence of a vagina. If a urethroscope is used to examine the vagina, a cervix can be seen. A catheter inserted through the cervix can be used for uterus and fallopian tube imaging.
(3) Generation and transformation tests show excessive excretion of steroids in the urine.
Female pseudohermaphroditism can also result from androgens transmitted through the placenta from the mother during pregnancy. These androgens may originate from virilizing ovarian tumors or from the mother’s treatment with corticosteroids or synthetic progestins during pregnancy.
2. In males: The cell nucleus is chromatin-negative, the genetic type is XY, and there is increased excretion of steroid substances in the urine, similar to females.
3. Differential diagnosis between hyperplasia and tumor: This is often difficult. A large tumor may be palpable, but bilateral adrenal hyperplasia can also be palpable shortly after birth. The dexamethasone suppression test for the hypothalamus may be helpful. Measurement of urinary steroids aids in diagnosing carcinoma, hyperplasia, or adenoma. The greatest difficulty is that urinary 11-OXOS excretion may be normal in cases of tumor. Fortunately, at least one steroid value is elevated. X-ray examinations can sometimes be helpful. If a tumor cannot be ruled out after multiple examinations, surgical exploration is necessary.
bubble_chart Treatment Measures
1. Treatment of Virilizing Hyperplasia: For congenital or acquired virilizing hyperplasia, hydrocortisone (hydrocortisone) can be used. Hydrocortisone suppresses the production of ACTH, thereby reducing the secretion of androgens while also supplementing corticosteroids, which are deficient. Steroid therapy should not be used rashly for mild hirsutism or minor steroid metabolic abnormalities, as it may pose other risks and has little effect on hair growth. However, it is highly valuable for congenital hyperplasia in children and adult sexual dysfunction. A relatively large dose is initially administered to achieve sufficient adrenal suppression, followed by gradual reduction to a maintenance dose. In growing children, adjusting treatment in relation to skeletal development is crucial. Insufficient suppression leads to rapid growth but ultimately short stature, while excessive suppression unnecessarily hinders growth. A growth chart should be maintained, with periodic skeletal X-rays to ensure normal development.
Hydrocortisone can be administered orally or by injection. Oral administration is preferable, as injections cause discomfort in children and inconvenience in adults. Oral hydrocortisone is taken 3–4 times daily for rapid effect. The oral dose is 2–3 times the intramuscular dose. For adults, the suppressive dose is 100 mg/day. For children under two years old, 20–30 mg/day achieves suppression within a few days, as confirmed by urinary 17-OXOS levels (refer to the table below):
Table 2: Normal Values of 24-Hour Urinary 17-OXOS
| Adults | 8–12 mg/24-hour urine |
| Children 6–14 years | 4–6 mg/24-hour urine |
| 2–6 years | 2–4 mg/24-hour urine |
| Infants under 2 years | 0.5–1 mg/24-hour urine |
Maintenance doses vary by patient. Adults typically require 30–40 mg/day, while children need 10–20 mg/day. Synthetic steroids may require smaller doses, but hydrocortisone alone or combined with other synthetic drugs is preferred. To ensure optimal ACTH suppression at night, prednisolone (5 or 7.5 mg) is often added at 11 PM, followed by 2.5 mg at 7 AM and 3 PM, with adjustments as needed—especially critical for growing children. Regular monitoring of 24-hour urinary 17-OXOS is essential. This treatment is vital for life.
For infants with Grade I sodium-losing congenital adrenal hyperplasia, hydrocortisone alone may maintain electrolyte balance, but 2–5 g of salt monthly is often required. Severe cases may need additional fludrocortisone (0.05–0.1 mg). Other salt-retaining hormones, such as intramuscular DOCA (125 mg pellet every 9–12 months) or synthetic desoxycorticosterone pivalate (2 mg every 3–4 weeks), can also be used. Salt-retaining hormone therapy should continue until age 3–4, then gradually taper off.
For adults after puberty with mild hyperplasia and sexual dysfunction, synthetic hormones can be used for treatment. Taking prednisolone 5 or 7.5 mg at 11 p.m. daily is sufficient.
Patients undergoing long-term steroid suppression therapy have poor emergency response capabilities and may develop acute adrenal cortical insufficiency. Therefore, when they become ill or undergo surgery, additional hydrocortisone supplementation is required. In untreated cases, where adrenal insufficiency remains undetected until adulthood, a severe crisis may occur during minor surgery.
Treatment Outcomes
For those who receive hormone therapy before the age of two, the results are highly satisfactory. With proper suppression therapy maintained, growth and development proceed normally. For treatment initiated after the age of two, periodic examinations show that skeletal development and final height depend on the timing of treatment initiation. Therapy can also resolve issues of virilization.
In females, post-treatment feminization occurs rapidly, with breast enlargement and normal menstruation. If treatment begins after puberty, further height increase becomes difficult. Hirsutism may gradually diminish, but if it was very pronounced before treatment, complete resolution may not occur. In such cases, cosmetic treatments can be applied. Normal sexual function and fertility in adulthood can be ensured, with breast enlargement, though a low-pitched voice and excessive hair may persist. Normal pregnancy is possible, and hydrocortisone is not required except during childbirth.
In boys, treatment prevents genital overdevelopment, improves growth, and allows the testes to mature normally after puberty. Hydrocortisone therapy can continue until fertility is achieved, while monitoring for adrenal insufficiency.
Surgical and cosmetic treatment: Correcting female external genital pseudohermaphroditism should be done as early as possible. Problems may arise in those raised as boys, often due to delayed appropriate treatment.
2. Treatment of Adrenal Tumors
Tumors should be surgically removed whenever possible. Excision of an adenoma often leads to full recovery, though hirsutism may persist for some time. Carcinomas may be difficult to resect or may recur quickly after removal. Chemotherapy with O,P′-DDD can provide temporary improvement, but the prognosis is poor.
Feminizing tumors are extremely rare and result from estrogen-secreting tumors of the adrenal gland or ectopic adrenal tissue. These tumors are often malignant, large, and palpable. They typically occur in adults, usually males. Histologically, these tumors are indistinguishable from other hormone-secreting adrenal tumors.
Clinical manifestations arise from excessive estrogen or gonadotropin secretion, suppressing pituitary gonadotropin production, leading to testicular atrophy, loss of libido, impotence, breast enlargement with tenderness (though nipple discharge is rare), fine facial skin, reduced facial hair, possible weight gain, and poor muscle development.
Surgical removal is preferred when possible. If resection is unfeasible or the tumor recurs, chemotherapy with O,P′-DDD is used. Hydrocortisone therapy is unnecessary. The prognosis remains poor.
If the excised tumor is benign, a cure is achieved. However, breast enlargement may not fully regress and may require cosmetic treatment.


