| disease | Acute Pancreatitis (Surgery) |
| alias | Acute Pancreatitis, AP |
Acute pancreatitis is a common condition in abdominal surgery, and the incidence of severe pancreatitis has been gradually increasing in recent years. Due to its significant physiological disturbances and pronounced damage to major organs, the mortality rate is very high. It can sometimes lead to sudden death. The mortality rate for severe pancreatitis is 20%, and it can rise to 50% in cases with complications. Clinically, acute pancreatitis is often classified into edematous and hemorrhagic necrotic types. While this classification reflects the pathological conditions, the progression of pancreatitis is not static. The pathological changes develop dynamically depending on the degree of pancreatic duct obstruction and alterations in the interstitial blood vessels (arteries, veins, and lymphatic vessels). Therefore, classifying acute pancreatitis into mild and severe types is more suitable for clinical application. Although clinicians often pay close attention to severe pancreatitis (such as hemorrhagic necrotic type), mild pancreatitis (such as edematous type) should not be overlooked, as it can progress to severe pancreatitis.
bubble_chart Etiology
The etiology of this disease remains incompletely understood, primarily due to significant differences between animal models and clinical observations. Current data suggest that the etiology of pancreatitis is associated with the following factors.
1. Obstructive factors
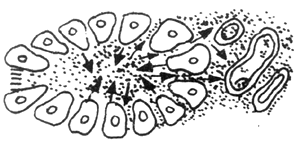
Conditions such as biliary ascariasis, impacted stones at the ampulla of Vater, and stenosis of the duodenal papilla can lead to bile reflux (Figure 1). However, this phenomenon cannot explain: ① Approximately 30% of cases where the biliary and pancreatic ducts do not share a common channel; ② Cases where autopsy reveals no abnormalities in the biliary tract and no history of alcoholism, yet acute pancreatitis occurs; ③ After pancreatic duct ligation, chronic pancreatitis generally develops, while acute pancreatitis is rare. Therefore, it is believed that under normal circumstances, bile reflux into the pancreatic duct does not cause pancreatitis unless a high-pressure environment is created within the pancreatic duct. For instance, significant obstruction at the lower end of the bile duct can lead to high biliary pressure, forcing bile under high pressure to reflux into the pancreatic duct, rupturing pancreatic acini and allowing pancreatic enzymes to enter the pancreatic interstitium, thereby causing pancreatitis. Animal experiments have shown that low-pressure perfusion of the pancreatic duct does not induce acute pancreatitis, whereas excessive pressure does. Clinically, it has been observed that excessive perfusion pressure during ERCP can lead to acute pancreatitis. Electron microscopy of the pancreatic duct after high-pressure perfusion reveals rupture at the junction of the pancreatic ductules and acini, with subsequent infiltration along the cytoplasmic and basement membranes, ultimately destroying the basement membrane and spreading into the connective tissue. If biliary stones do not cause ampullary obstruction or high-pressure bile reflux into the pancreatic duct, it should not be classified as biliary acute pancreatitis.
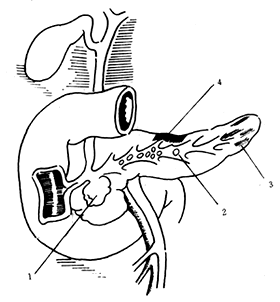
⑴
⑵
Figure 1 Obstructive factors in pancreatitis
⑴⒈ Pseudocyst ⒉ Pancreatic stones ⒊ Fibrosis ⒋ Tissue degeneration
⑵⒈ Sphincter spasm ⒉ Fibrous edema ⒊ Stones ⒋ Tumor ⒌ Primary cyst
Analysis of a large number of intraoperative cholangiograms has revealed that patients with a history of pancreatitis exhibit certain anatomical features of the biliary and pancreatic ducts, such as a wider pancreatic duct, a larger angle between the biliary and pancreatic ducts, a longer common channel, and a wider cystic and common bile duct, along with numerous small and irregularly shaped stones. These characteristics of emetic therapy facilitate the migration of small gallstones into the common bile duct, where they may temporarily lodge and obstruct the ampulla, leading to pancreatitis.
Sometimes, when stones pass through the Oddi sphincter (especially irregularly shaped ones), they can cause sphincter spasm, resulting in high-pressure bile reflux into the pancreatic duct and triggering pancreatitis. During biliary infections, bacteria can travel through the common lymphatic vessels of the bile and pancreas back into the pancreas, and if the Oddi sphincter is stenotic to varying degrees, pancreatitis can occur.
Does the presence of bacteria in the pancreas lead to pancreatitis? Widdison et al. conducted animal experiments as follows: They induced experimental pancreatitis by perfusing pancreatic enzymes through the main pancreatic duct. The animals were divided into five groups: Group I received tracer Escherichia coli, while Groups II–V were injected with a certain amount of E. coli via the gallbladder, main pancreatic duct, obstructed renal pelvis, or colon, respectively. Pancreatic cultures were performed after 24 hours, except for the colon group, which was cultured after 72 hours. All groups showed E. coli growth in the pancreas. Compared to the group with chronic bacterial colonization, except for the colon group, bacterial growth in pancreatitis was more frequent, and bacteria were often found in the pancreas, but this did not necessarily lead to infectious pancreatitis.
2. Alcohol factorsChronic alcohol consumption is prone to cause pancreatitis, which is a common phenomenon in Western countries, accounting for 70%. The pathogenesis of alcoholic pancreatitis includes: ① Alcohol stimulates the parietal cells of the stomach to produce a large amount of gastric acid, which then reaches the duodenum and stimulates the S cells and small intestine wall's I cells, leading to the production of CCK-PZ. This creates a high-pressure environment in the pancreatic duct within a short period. ② Frequent alcohol stimulation of the duodenal wall causes congestion and edema, which may extend to the duodenal papilla, resulting in relative obstruction of the biliary and pancreatic duct openings. ③ Long-term alcohol consumption increases protein secretion in the pancreatic duct, forming "intraductal protein plugs," which obstruct the pancreatic duct. Under these conditions, excessive alcohol consumption and overeating can promote the massive secretion of pancreatic enzymes, causing a sudden rise in pancreatic duct pressure. This leads to the rupture of pancreatic acini, allowing pancreatic enzymes to enter the interstitial space between acini and triggering acute pancreatitis. The simultaneous intake of alcohol with high-protein and high-fat meals not only increases pancreatic enzyme secretion but also induces hyperlipoproteinemia. At this point, pancreatic lipase breaks down triglycerides, releasing free fatty acids that damage the pancreas.
The analysis of domestic and international disease causes of acute pancreatitis is listed below (Table 1, Table 2).
Table 1 Analysis of disease causes of acute pancreatitis in China△
| Author | Time | Region | Number of cases | Alcohol% | Biliary disease% | Overeating% |
| Zheng Xianli | 1975 | Tianjin | 820 | - | 16.83 | - |
| Shen Jie | 1979 | Qingdao | 81 | 2.5 | 35.8 | 30.9 |
| Huang Zhiqiang | 1980 | Chongqing | 618 | 9.2 | 20.8 | 3.4 |
| Qian Yunqing | 1984 | Shanghai | 71 | - | 20 | 20 |
△Excerpted from Qian Li, Modern General Surgery, 1993.404
Table 2 International disease causes of acute pancreatitis (Ranson 1983)
| Author | Time | Number of patients | Related disease causes% | ||
| Gallstones | Alcohol-related | Others | |||
| Gliedman | 1970 | 2066 | 15 | 75 | 10 |
| Gillespie | 1973 | 93 | 34 | 23 | 43 |
| Olsen | 1974 | 100 | 12 | 87 | 16 |
| Howes | 1975 | 95 | 5 | 91 | 4 |
| Trapnell | 1975 | 590 | 54 | 4 | 42 |
| Jacobs | 1976 | 543 | 44 | 30 | 34 |
| Wellbourn | 1977 | 257 | 50 | 9 | 41 |
| Imrie | 1978 | 161 | 52 | 32 | 17 |
| Ranson | 1978 | 450 | 16 | 70 | 14 |
| Madsen | 1979 | 122 | 14 | 40 | 38 |
| Stiani | 1979 | 389 | 3 | 92 | 5 |
| Kelly | 1980 | 275 | 63 | 24 | 13 |
3. Vascular Factors
Acute embolism or obstruction of the small arteries and veins in the pancreas can lead to acute circulatory disturbances, resulting in acute pancreatitis. This phenomenon has been confirmed. Experimental studies have shown that injecting microspheres with diameters of 8–20 μm into the pancreas can induce significant experimental pancreatitis. During autopsies of pancreatitis cases, Popper observed atherosclerotic thrombi in the pancreatic vessels.
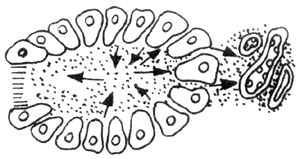
Another vascular factor is based on pancreatic duct obstruction. When the pancreatic duct is obstructed, the high pressure within the duct causes pancreatic enzymes to be passively "leaked" into the interstitial tissue. The stimulation from these enzymes leads to thrombosis in the lymphatic vessels, veins, and small arteries within the interstitial tissue, subsequently causing pancreatic ischemia and necrosis. The progression of this process is illustrated in Figure 2. As shown in the figure, there is no clear boundary between edematous pancreatitis and hemorrhagic necrotizing pancreatitis. If not managed properly, the former can develop into the latter.
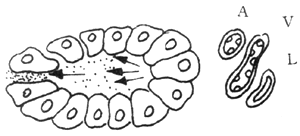
1
2
3
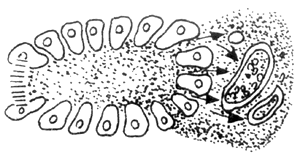
4

5
Figure 2 Vascular Factors
A: Pancreatic interstitial stirred pulse V: Pancreatic interstitial vein L: Pancreatic interstitial lymphatic vessel
1. Normal pancreatic acini, pancreatic enzymes drain into small pancreatic ducts
2. After pancreatic duct obstruction, pancreatic enzymes reflux along acinar cells into the pancreatic interstitium, stimulating arteries, veins, and lymphatic vessels
3–5. As the degree of pancreatic duct obstruction worsens, the reflux of pancreatic enzymes into the interstitium increases, and the arteries, veins, and lymphatic vessels eventually become completely occluded
Reily conducted experiments on the fundamental hemodynamic mechanisms of pancreatic ischemia in cardiogenic shock. By inducing pericardial tamponade in pigs to create cardiogenic shock and observing pancreatic hemodynamics, it was found that the uneven reduction in pancreatic blood flow was more pronounced than the decrease in cardiac output in animals with shock alone. The increase in pancreatic ischemia was caused by selective pancreatic vasoconstriction and reduced perfusion pressure. Pancreatic ischemia occurring during cardiogenic shock is primarily due to selective pancreatic vasoconstriction.
Castillo et al. studied 300 patients undergoing extracorporeal circulation surgery to observe whether extracorporeal circulation caused injury to the pancreas. Blood amylase, pancreatic isoenzymes, and lipase were measured on postoperative days 1, 2, 3, 7, and 10. Among 80 cases, 23 had abdominal findings, 3 developed severe pancreatitis, and 11% died postoperatively from secondary pancreatitis.
4. Trauma and Iatrogenic Factors
Pancreatic trauma causes pancreatic duct rupture, pancreatic fluid leakage, and insufficient blood supply after injury, leading to the development of severe acute pancreatitis. Iatrogenic pancreatitis can occur in two scenarios: one occurs during gastrectomy, particularly when there is a gastric antrum flush or a duodenal posterior wall ulcer penetrating the pancreas. During gastrectomy, scraping the ulcer surface on the pancreas can form a pancreatic fistula, leading to pancreatic fluid leakage and autodigestion of the pancreas. The other scenario is postoperative pancreatitis without direct pancreatic involvement, often caused by surgery on adjacent organs. This may result from Oddi's sphincter edema, impaired pancreatic fluid drainage, injury to pancreatic blood supply, or various stimuli to the vagus nerve causing excessive pancreatic secretion.
5. Infectious Factors
Acute pancreatitis can occur due to various bacterial and viral infections, such as mumps virus, adenovirus, hepatitis A virus, and bacterial pneumonia. Viruses or bacteria enter pancreatic tissue through the blood or lymph, causing pancreatitis. Generally, such infections result in simple edematous pancreatitis, with hemorrhagic necrotizing pancreatitis being less common.
6. Metabolic Disorders
(1) Hypercalcemia: Hypercalcemia-induced pancreatitis may be related to the following factors: calcium salt deposition forming calcifications in the pancreatic ducts, blocking the ducts and causing pancreatic fluid to enter the interstitium, leading to pancreatitis; promoting pancreatic secretion; and converting trypsinogen into trypsin.
(2) Hyperlipidemia: About one-fourth of acute pancreatitis cases involve hyperlipidemia. This may be due to small pancreatic vessels being occluded by aggregated serum lipid particles, or high concentrations of pancreatic lipase breaking down serum triglycerides, releasing large amounts of free fatty acids that damage and occlude small pancreatic vessels. Pancreatitis can occur when serum triglycerides reach 5–12 mmol/L.
7. Other Factors
Such as drug allergies, drug poisoning, hemochromatosis, adrenal corticosteroids, and genetics.
The pathogenesis of acute pancreatitis (AP) primarily involves the self-digestion of the pancreas by pancreatic enzymes and the digestion of surrounding tissues, leading to a series of secondary organ dysfunctions. The pancreas contains a rich variety of digestive enzymes: proteases, lipases, amylases, etc. The enzymes secreted by pancreatic acinar cells mainly include trypsin, chymotrypsin, carboxypeptidase, elastase, phospholipase A2, hard protease, lipase, amylase, nucleoprotease, etc. Under normal circumstances, except for lipase, amylase, and nucleoprotease, which exist in active forms, the others are in inactive states. Under pathological conditions, these enzymes become activated within the pancreatic ducts and cells, triggering the onset of pancreatitis.
Activation of pancreatic enzymes within the pancreatic ducts:
Due to various factors, such as bile, duodenal fluid, intestinal enzymes, emulsified fats, and lysolecithin refluxing into the pancreatic ducts, the proenzymes within the ducts become activated. The activated enzymes then digest the pancreatic tissue, leading to pancreatitis.
Activation of pancreatic enzymes within cells:
The proenzyme granules within pancreatic acinar cells contain pancreatic secretory trypsin inhibitor (PSTI), which prevents intracellular enzyme activation. Under normal conditions, a lysosomal enzyme formed within the cells remains separate from the enzyme granules. Under the influence of pathogenic factors, the enzyme granules and lysosomes fuse through a phagocytic phenomenon. At low pH levels, this leads to intracellular activation of proenzymes, damaging the cells themselves. If pancreatic enzymes leak into the interstitial tissue, the pancreatic lesions worsen and cause damage to adjacent organs. Continued progression of the lesions can result in multi-organ injury.
In addition to the aforementioned self-digestion mechanism, recent in-depth research on acute pancreatitis has identified other critical factors in its pathogenesis, including the trypsin-antitrypsin system, phospholipase A and thromboxane A2, pancreatic circulatory disorders, oxygen free radicals, cell membrane stability, and endotoxins.
1. Trypsin-Antitrypsin System
The pancreas secretes various antitrypsin factors that inhibit the self-activation and self-digestion of trypsin. In severe pancreatitis, local antitrypsin is depleted, leading to uncontrolled activation of pancreatic enzymes and self-digestion. Mesotrypsin (MT), which is not inhibited by antitrypsin, constitutes about 10% of the trypsin in pancreatic juice but is three times more active than trypsin. It counteracts trypsin inhibitors, posing a severe threat to pancreatic integrity and extra-pancreatic tissue damage.
Experimental evidence shows that antitrypsin therapy is significantly effective in treating acute pancreatitis. Freeze-dried plasma contains large amounts of antitrypsin, so its use in acute pancreatitis patients not only replenishes colloids but also provides antitrypsin.
2. Phospholipase A and Thromboxane A 2
Phospholipase A (PLA) is referred to as the "key enzyme" in the pathogenesis of acute pancreatitis. The self-digestion of pancreatic acinar cells is directly related to PLA. At the onset of acute pancreatitis, PLA proenzyme is activated by bile salts, trypsin, calcium ions, and enteropeptidase. Subsequently, PLA hydrolyzes the lecithin in the cell membrane, generating free fatty acids (FFA) and lysolecithin. The latter can disrupt the cell membrane, releasing various intracellular digestive enzymes, leading to pancreatic hemorrhage, necrosis, and damage to other organs. In acute pancreatitis patients, PLA exists in two distinct forms in the plasma.
When PLA decomposes FFA and lysophosphatidylcholine from the membrane phospholipid, it produces a thromboxane A2 that causes strong vasoconstriction. When thromboxane A2 (TXA2) pathologically increases, and the imbalance of TXA2/PGI2 occurs, it can lead to impaired blood supply to the pancreas, while also damaging the lysosome membrane within cells and increasing intracellular calcium ions. By preventing the increase of TXA2 and maintaining the balance of TXA2/PGI2, the progression of acute pancreatitis can be effectively controlled.
3. Lysosomal Enzymes
Previously, it was believed that the activation of pancreatic enzymes during acute pancreatitis occurred extracellularly in the acinar cells. However, recent studies have found that the activation of zymogens can occur intracellularly in the acinar cells through the action of lysosomal hydrolases. The lower pH in this environment can inactivate pancreatic trypsin inhibitors, leading to the intracellular activation of pancreatic enzymes. Some researchers suggest that the activation of trypsinogen by lysosomes within the acinar cells is a critical step in pancreatic autodigestion and hemorrhage. Wilson's study indicates that lysosomal involvement is a significant factor in alcoholic acute pancreatitis.
4. Pancreatic Microcirculatory Disturbance
During pancreatitis, pancreatic blood flow decreases, which is distinctly different from other tissues. Moreover, pancreatic tissue is highly sensitive to changes in blood flow. In severe acute pancreatitis (acute hemorrhagic pancreatitis), pancreatic blood flow is significantly reduced. In acute edematous pancreatitis, progressive ischemia in pancreatic tissue, if not alleviated, can lead to acute hemorrhagic pancreatitis. The underlying cause is vascular thrombosis or obstruction between pancreatic acini, resulting in ischemia, necrosis, and progressive worsening of the condition. Another factor contributing to pancreatic microcirculatory disturbance is the inflammatory stimulation of capillaries, particularly the occlusion of small veins, which further impairs venous return and exacerbates the disease. Studies suggest that circulatory disturbances play a crucial role in the pathogenesis of alcoholic pancreatitis. Ssafey et al. propose that increased capillary permeability is a key pathophysiological phenomenon in the early stages of acute pancreatitis. Therefore, improving capillary permeability could be highly beneficial in treating acute pancreatitis. Capillary permeability is closely related to oxygen free radicals. Ven Ooijen's experiments indicate that the increase in TXA2 is a critical factor in ischemia-induced acute pancreatitis. Ischemia and hypoxia activate platelets, and their aggregation, along with the action of TXA2, further aggravates pancreatic tissue ischemia.
5. Oxygen-Derived Free Radicals
Recent studies suggest that oxygen free radicals are involved in the pathophysiological process of acute pancreatitis. These radicals can be scavenged by endogenous superoxide dismutase (SOD) and catalase (CAT).
Superoxide Dismutase (SOD): Present in the cytosol and mitochondria, SOD is a specific enzyme that scavenges reactive oxygen species (ROS) in the body. It accelerates the dismutation of superoxide radicals, neutralizing ROS produced during normal metabolism.
Catalase (CAT): It catalyzes the reduction of H2O2 to H2O and serves as an important cellular scavenger of oxygen free radicals.
Glutathione Peroxidase: Found in the cytoplasm and mitochondria, it participates in the reduction of various peroxides.
Under normal physiological conditions, oxygen free radicals and the scavenging system are in balance. When the function of oxygen free radicals and the scavenging system declines, it leads to damage to the pancreas by reactive oxygen species. Oxygen free radicals can cause damage to macromolecules such as proteins, nucleic acids, lipids, and polysaccharides, thereby increasing the capillary permeability of the pancreas, resulting in pancreatic edema, hemorrhage, tissue degeneration, and necrosis. In acute pancreatitis, the SOD (superoxide dismutase) levels in pancreatic tissue decrease, while the SOD activity in the blood increases, which is caused by the rise in lipid peroxides due to oxygen free radicals. Oxygen free radicals can also reduce membrane stability, leading to the release of lysosomes from pancreatic acinar cells and the activation and release of various pancreatic enzymes. Additionally, oxygen free radicals can activate phospholipase A, causing the breakdown of lecithin on pancreatic cell membranes and further injuring pancreatic tissue. Some researchers have administered SOD and CAT (catalase) via intravenous injection, but their active duration was very short, lasting only a few minutes. Therefore, others have combined these two enzymes with a macromolecular polymer, polyethylene glycol (PGE), which allows them to maintain activity in plasma for 30–40 hours, significantly alleviating pancreatitis in rats.
6. Others
Internal toxinemia is also involved in the development of acute pancreatitis. It is caused by the internal toxinemia produced during acute pancreatitis, which in turn exacerbates pancreatic injury. Some believe that internal toxins damage mitochondrial structure, affect ATPase and oxidative phosphorylation coupling processes, leading to energy metabolism disorders; alter the body's immune function; directly destroy lysosomal membranes within mononuclear phagocyte system cells, thereby causing cellular damage; and can induce a series of pathological changes in the body: vasomotor dysfunction, decreased platelets and white blood cells, etc.
In summary, the pathogenesis of acute pancreatitis is complex, as the actions of various enzymes can have both positive and negative effects on pancreatic cell membranes and organelles. Currently, research is in a phase of in-depth study. It is firmly believed that the onset of acute pancreatitis is often not due to a single mechanism but rather the result of multiple factors mutually reinforcing each other, forming a vicious cycle. If this chain can be effectively broken, the treatment of acute pancreatitis will achieve a significant breakthrough.
The clinical pathophysiology of severe acute pancreatitis is summarized below (Table 1). 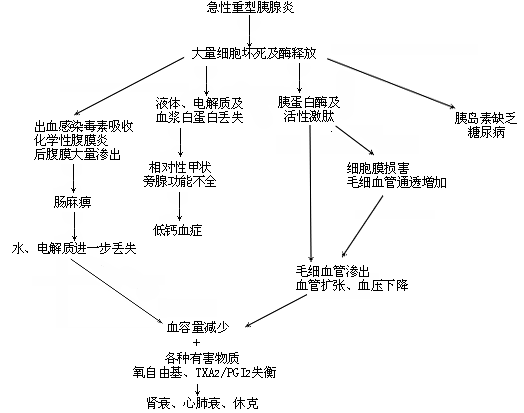
bubble_chart Pathological Changes
(1) Substances secreted by the pancreas
Secretin: It is a polypeptide composed of 25 amino acids. Its secretory cells are distributed in the duodenum and the upper jejunum. When the pH in the lumen of the small intestine decreases, it stimulates an increase in secretin secretion. Secretin increases HCO3- secretion, and the increase is dose-dependent. Large doses of secretin can inhibit the secretion of pancreatic enzymes. In chronic pancreatitis, the pancreas's response to secretin stimulation is reduced.
Vasoactive intestinal peptide (VIP): A polypeptide extracted from the small intestinal mucosa with strong vasodilatory effects. Its molecular structure is similar to that of secretin. Cells secreting VIP are found throughout the intestinal tract, with the highest concentration in the ileum. It is also present in the nerve endings of the intestinal wall, leading to the belief that it also functions as a neurotransmitter.
CCK-PZ: A peptide substance extracted from the duodenal and jejunal mucosa. It stimulates the pancreas to secrete large amounts of pancreatic juice, known as pancreozymin. It is identical to cholecystokinin (CCK) and has both gallbladder-contracting and sphincter-relaxing effects, hence it is called cholecystokinin-pancreozymin (CCK-PZ).
Substances that inhibit pancreatic secretion
Glucagon: It inhibits the secretion of HCO3- stimulated by secretin. The inhibitory effect is dose-dependent. It also competitively inhibits the secretion of pancreatic enzymes stimulated by CCK.
Calcitonin: It inhibits pancreatic secretion stimulated by secretin or CCK. Its effect is not accompanied by a decrease in blood calcium, so it is not due to hypocalcemia.
Epinephrine and norepinephrine: These are drugs that stimulate α-adrenergic receptors. They reduce secretion in the normal or stimulated pancreas by causing intense vasoconstriction. However, if the vasoconstrictive effect is blocked by α-adrenergic receptor inhibitors, norepinephrine instead increases the secretion of water and electrolytes by the pancreas.
Anticholinergic drugs: Such as atropine, which can reduce pancreatic secretion. Their effect is more pronounced in counteracting vagal nerve excitation than in opposing direct CCK stimulation.
(2) Effects of acute pancreatitis on various body systems
1. Acute pancreatic pulmonary failure
The impact of acute pancreatitis, especially severe acute pancreatitis, on respiratory function has been noted by clinicians for decades. The main manifestations include dyspnea and cyanosis, but these symptoms do not always correlate with the severity of pancreatitis. Since the 1970s, understanding has deepened, confirming it as adult respiratory distress syndrome (ARDS).
ARDS is a common and severe complication of severe acute pancreatitis. According to statistics, progressive dyspnea occurs in 14.2% to 33% of patients with severe acute pancreatitis (more common in first-time cases). When dyspnea appears, the mortality rate can be as high as 30% to 40%. In 1974, Feller reported 200 cases of acute pancreatitis, among which 83 were severe cases, 18 had respiratory failure, and 17 (without oxygen inhalation) showed a stirred pulse with arterial oxygen partial pressure below 9.33 kPa, accounting for 38%. Early pulmonary function tests after onset revealed decreased inspiratory capacity, increased resistance, and reduced pulmonary diffusion capacity. Observations from experimental acute pancreatitis demonstrated increased arteriovenous shunting, as well as decreased minute respiratory volume, oxygen consumption, and pulmonary stability index. Acute respiratory insufficiency can occur in the early stages of acute pancreatitis, with stirred pulse hypoxemia being a common early symptom, which may appear before any changes are visible on chest X-rays. In 1974, Olsen reported a case series (100 cases), with 80 undergoing chest X-ray examinations, of which 25% showed pleural effusion, atelectasis, pneumonia, etc.
(1) Acute respiratory distress syndrome (ARDS) caused by acute pancreatitis has many underlying causes, and there is no definitive conclusion yet. The current views of some scholars are summarized as follows:
A. The role of pancreatic enzymes: Carbohydrate enzymes seem harmless to tissues, while protein and lipid enzymes are significant pathogenic factors. Trypsin includes trypsinogen, chymotrypsinogen, procarboxypeptidase, aminopeptidase, proelastase, collagenase, etc. Among these, trypsinogen and proelastase play the most critical roles. Activated trypsin (Trypsin) can activate almost all pancreatic enzymes and also activate Factor VII, subsequently triggering multiple enzyme systems such as coagulation, fibrinolysis, complement, and kinin. Trypsin (Try) is an endopeptidase, and human Try can be divided into Try-I and Try-II. Try-I is a cationic protein, while Try-II is an anionic protein. The normal blood level of Try is about 300–460 ng/dl, but it can increase more than 10-fold during acute pancreatitis. Elastase plays a major role in pulmonary hemorrhage and edema, causing damage to vascular walls. It also hydrolyzes elastic fibers and acts on various other protein substrates, such as hemoglobin, casein, fibrin, and albumin.
Lipases include lipase, colipase, cholesterol esterase, and prophospholipase A. The first three primarily hydrolyze their respective substrates to produce free fatty acids (FFA), which not only cause tissue injury but are also cytotoxic, leading to cell degeneration, necrosis, and lysis, with significant damage to the lungs. Phospholipase A (PLA) can also decompose lecithin to produce FFA and lysolecithin. PLA can be activated by Try and is classified into PLA1 and PLA2, with the latter being more stable. Generally, the PLA referred to is PLA2.
The effects of PLA include: hydrolyzing lecithin to produce FFA and lysolecithin; hydrolyzing pulmonary surfactant, leading to atelectasis; hydrolyzing phospholipids in cell membranes, affecting membrane permeability; hydrolyzing phospholipid-containing enzymes in mitochondrial membranes, thereby disrupting oxidative phosphorylation; and reducing the stability of lysosomes in lung cells, causing their release and subsequent tissue damage, resulting in abnormal lung perfusion.
Activated Try can trigger multiple enzyme systems in the blood, altering blood viscosity. Insufficient lung perfusion reduces lung function, decreases surfactant synthesis, and leads to the accumulation of metabolic byproducts locally. Damaged pulmonary vessels exhibit increased permeability. In acute pancreatitis, cardiac output is often increased, while peripheral vascular resistance decreases, creating a hyperdynamic circulatory state, which may be related to a significant increase in pulmonary arteriovenous shunting. Some lung tissues experience underperfusion, while others are overperfused, which may be another characteristic of pancreatic-induced lung injury.
B. The role of the coagulation system: During acute pancreatitis, Try is released into the bloodstream, activating the coagulation system and causing pulmonary thrombosis and embolism. On the basis of embolism, vasoactive substances such as histamine and serotonin are released, leading to pulmonary vasoconstriction, vascular membrane injury, increased permeability, and the formation of pulmonary edema.
C. The role of the complement system: Complement is a group of immunoglobulin molecules in the blood. Once activated, it triggers a chain reaction, generating many active fragments and complex molecules that cause multifaceted injury effects. For example, C3a, C5a, C567 can induce perivascular mast cells to release histamine, leading to vascular dysfunction and endothelial membrane injury. Complement (C3) is activated by Try, and C3 can also be indirectly activated through the activation of factor VII. The injury caused by complement is systemic, and lung injury is no exception.
In addition, kinin substances can also cause lung damage and vasodilation after activation by Try, injuring the vascular endothelium and increasing vascular permeability.
In recent years, the role of free radicals in lung damage caused by pancreatitis has also attracted much attention. For example, O2-, H2O2, and OH-, which are products of peroxidation and phospholipid breakdown, can cause vascular dysfunction, endothelial dysfunction, increased permeability, bronchial smooth muscle contraction, and mucosal edema.
In other cases, severe dehydration during acute pancreatitis leads to a hypercoagulable state, often accompanied by endotheliitis, causing platelet, leukocyte, and erythrocyte aggregation that blocks microvessels. Some patients may experience severe abdominal pain, leading to pulmonary arteriolar spasm due to neural reflexes and the action of substances like catecholamines and histamine. Histamine not only contracts arterioles but also venules. Other factors, such as abdominal distension, diaphragmatic elevation, and pleural effusion in acute pancreatitis, can also impair respiration. In rare cases, increased fibrinogen in acute pancreatitis forms abdominal masses in the lungs, severely disrupting gas exchange.
(2) When pulmonary failure occurs, the lungs exhibit increased weight, consolidation, and scattered multiple hemorrhagic spots. Microscopic examination reveals interstitial congestion, edema, and intra-alveolar and extra-alveolar hemorrhage. Focal alveolar collapse and fusion may also be observed. Functional residual capacity decreases, the dead space-to-tidal volume ratio increases, pulmonary shunting worsens, and diffusion capacity declines. Hyperventilation leads to respiratory alkalosis. As lactic acidosis worsens and CO2 elimination is impaired, respiratory and metabolic acidosis may eventually develop.
2. Cardiovascular Changes in Severe Acute Pancreatitis
Sudden death cases in severe pancreatitis often show significant cardiac damage upon autopsy, such as myocardial infarction, endocarditis, or conduction system impairment.
Example: A 58-year-old male patient, Hou ×, experienced insomnia and loss of appetite for three weeks but no abdominal pain or fever and remained able to work. One night, he felt fatigued but fell asleep. By 5 AM the next day, he was found dead. Autopsy confirmed hemorrhagic necrotizing pancreatitis with minimal peritoneal effusion. Amylase levels exceeded 1064 units (Winslow units). Pericardial effusion and a large infarct in the anterior left ventricular wall were observed.
The mechanism of cardiovascular damage in acute pancreatitis remains unclear. Current theories include pancreatic enzymes entering circulation, causing coronary artery spasm; trypsin and polypeptides directly damaging the myocardium; and inflammatory exudate stimulating the celiac plexus, leading to reflex vasospasm. Some propose that the pancreas contains a myocardial depressant factor, as pancreatic homogenate injected into arteries inhibits myocardial oxygen use. Others suggest that acute pancreatitis releases substances that overexcite the cardiac conduction system, causing arrhythmias or ventricular fibrillation.
3. Renal Failure in Acute Pancreatitis
Renal failure is common, occurring not only in hypovolemic cases but also in those with normal blood volume. The cause lies in protein breakdown products from pancreatic enzymes, which are nephrotoxic, damaging glomeruli and tubules, leading to epithelial swelling, detachment, and necrosis. Fibrinogen may deposit in glomeruli, and endotoxins increase renal arteriolar resistance, reducing renal blood flow.
4. Neuropsychiatric Changes
This change manifests as phenomena such as delirium, trance, unconsciousness, and even mental disorders. Infections, poisoning, high fever, and long-term alcoholism are causes of psychiatric symptoms. In recent years, it has been discovered that during acute severe pancreatitis, large amounts of phospholipase A are produced, which has a strong affinity for the nervous system and damages nerves. Additionally, lysolecithin, derived from the lecithinase that decomposes brain cells, is a component of snake venom with potent neurotoxicity. In a small number of patients, increased pancreatic lipase in the bloodstream leads to intracranial fat necrosis, softening, or hemorrhage, resulting in pancreatic encephalopathy.
5. Changes in Electrolytes
In severe pancreatitis, lipase breaks down neutral fats into glycerol and fatty acids, the latter of which combine with calcium to form soap, leading to acute hypocalcemia, a phenomenon well recognized. Additionally, during acute severe pancreatitis, glucagon is released, which stimulates the thyroid to secrete calcitonin, thereby inhibiting the parathyroid gland's ability to mobilize calcium from bones. However, administering glucagon to normal individuals does not induce hypocalcemia, leading some to currently believe that the cause of hypocalcemia is the breakdown of parathyroid hormone by proteases, preventing it from maintaining calcium levels. Both scenarios can be effectively addressed by administering parathyroid hormone.
bubble_chart Clinical Manifestations
The pathological changes of acute pancreatitis vary at different stages, and the systemic reactions also differ. Even for the same hemorrhagic necrotizing pancreatitis, there can be significant variations in clinical manifestations due to differences in the time of onset and the patient's condition. In general, the main symptoms of acute edematous pancreatitis are
abdominal pain, nausea, vomiting, and fever. In contrast, hemorrhagic necrotizing pancreatitis, in addition to the above symptoms, may also present with shock, high fever,
jaundice,
abdominal distension and fullness, intestinal paralysis, peritoneal irritation signs, and subcutaneous ecchymosis due to pancreatic hemorrhage, necrosis, and autolysis.
Abdominal pain is the earliest symptom, often occurring suddenly after overeating or extreme fatigue, typically localized in the upper abdomen, either centrally or slightly to the left. The pain is persistent, progressively worsening, and knife-like, radiating to the back and flanks. Severe abdominal pain is mostly caused by pancreatic edema or inflammatory exudate compressing and irritating the celiac plexus. In hemorrhagic necrotizing pancreatitis, the pain quickly becomes generalized, accompanied by acute
abdominal distension and fullness, as if air is being pumped into the abdomen, and varying degrees of shock may develop rapidly.
Nausea and vomiting are manifestations of vagus nerve irritation by inflammation. Initially, the vomitus contains food and bile-like substances. As the condition worsens (or in hemorrhagic necrotizing pancreatitis), intestinal paralysis occurs, and the vomitus becomes fecal-like.
Jaundice is less common in acute edematous pancreatitis, occurring in about one-fourth of cases, but more frequent in acute hemorrhagic pancreatitis. The causes of jaundice include concurrent biliary stone impaction, edema or spasm of the common bile duct orifice, compression of the distal common bile duct by an enlarged pancreatic head, or liver damage due to severe intra-abdominal infection.
Dehydration in acute pancreatitis is mainly caused by intestinal paralysis and vomiting in mild cases. In severe pancreatitis, severe dehydration and electrolyte imbalances can develop rapidly due to retroperitoneal inflammation, leading to the loss of several thousand milliliters of fluid into the retroperitoneal space, akin to invisible loss. In hemorrhagic necrotizing pancreatitis, severe dehydration, oliguria, or anuria may occur within hours to a dozen hours after onset.
Due to massive inflammatory exudation, pancreatic necrosis, and localized abscesses, varying degrees of fever may occur. Mild pancreatitis typically presents with a temperature below 39°C, subsiding within 3–5 days. In severe pancreatitis, the temperature often ranges between 39–40°C, accompanied by delirium lasting for weeks, along with signs of toxemia.
In rare cases of hemorrhagic necrotizing pancreatitis, pancreatic fluid and necrotic tissue can spread along tissue planes to the subcutaneous layer, dissolving subcutaneous fat and causing capillary rupture and hemorrhage. This results in localized bluish-purple discoloration of the skin, sometimes forming large patches. These may appear on the flanks (Grey-Turner sign) or around the umbilicus (Cullen sign).
The pancreas is deeply located. In mild edematous pancreatitis, deep tenderness is usually present in the upper abdomen, with occasional marked tenderness on the anterior abdominal wall. In severe acute pancreatitis, due to extensive pancreatic dissolution, necrosis, and hemorrhage, both the anterior and posterior peritoneum are affected, leading to generalized abdominal rigidity, tenderness, and distension. Large amounts of inflammatory
ascites may cause shifting dullness, and bowel sounds may disappear, indicating paralytic ileus.
Inflammatory stimulation from exudate can lead to reactive pleural effusion, more commonly on the left side, which may cause ipsilateral atelectasis and dyspnea.
When a large amount of necrotic tissue forms an
abdominal mass within the lesser omental sac, a raised lump may be visible in the upper abdomen, tender to palpation but often with indistinct borders. In some patients, abdominal tenderness and other signs may become less obvious, but persistent high fever, leukocytosis, and recurrent "partial intestinal obstruction"-like symptoms may indicate localized abscess formation in the abdominal or pelvic cavity. Ultrasound and digital rectal examination should be performed.
bubble_chart Auxiliary Examination
I. Laboratory Tests
(1) Amylase Test:
This traditional test method, though used for over half a century, remains a good, simple, and feasible means for diagnosing pancreatitis. Due to the retrograde flow of pancreatic enzymes into the blood or the reabsorption of exudate into the blood, amylase levels in blood and urine rise during acute pancreatitis. The normal value for blood amylase is <256 units by the Winslow method and <500 units by the Somogyi method. In mild acute pancreatitis, blood amylase rises within 6–12 hours after onset and gradually returns to normal within 48–72 hours, while urine amylase rises around 12–24 hours after onset and remains elevated for 3–5 days. However, in severe acute pancreatitis, the rise occurs earlier. Clinically, a comprehensive analysis of amylase level changes, combined with other symptoms, is necessary for an accurate diagnosis.
Normal amylase levels: This may indicate recovery, with the patient in good overall condition and no abdominal signs. In acute hemorrhagic necrotizing pancreatitis, if amylase levels do not rise during initial examination or treatment, it suggests progressive worsening of the condition due to extensive necrosis of pancreatic acinar cells, leading to a "depletion" of amylase secretion. This phenomenon, occasionally seen in acute hemorrhagic necrotizing pancreatitis, warrants high attention.
Elevated amylase levels: Some patients with seasonal diseases may exhibit abdominal pain and elevated amylase levels, but their clinical symptoms and signs do not indicate pancreatitis. Serum amylase testing is often nonspecific; acute pancreatitis can show varying degrees of elevation depending on severity. Other acute abdominal conditions, such as cholecystitis, cholelithiasis, biliary obstruction, intestinal obstruction, ulcer perforation (especially duodenal bulb perforation), mesenteric thrombosis, and morphine use, can also elevate amylase levels. In cholelithiasis, transient amylase elevation may occur due to stimulation of the Oddi sphincter during stone passage. In ulcer perforation, intestinal contents rich in pancreatic juice enter the peritoneal cavity, where amylase is absorbed, raising blood amylase levels. In intestinal obstruction, stagnant intestinal fluid allows amylase to seep into the peritoneal cavity through damaged intestinal walls and be absorbed. Thus, elevated amylase levels must be interpreted clinically and should not alone diagnose pancreatitis.
Macroamylasemia is a rare condition of unknown cause, possibly due to complexes of amylase and macromolecules in the blood that cannot be filtered by the glomeruli, leading to its accumulation. Its hallmark is high blood amylase but normal or low urine amylase, which may persist for months or even years.
Severe pancreatitis often involves large amounts of inflammatory ascites. Abdominal paracentesis should be performed to measure amylase levels, with the extracted ascites typically being bloody and turbid, often with high amylase content.
Robent et al. used pancreatic lipase and amylase levels in serum and ascites to diagnose necrotizing pancreatitis early. If both enzyme levels in ascites were higher than in blood, the Enzymatic Score (ES) was recorded as "2." If either enzyme level in ascites was higher than in blood, ES was "1." If both were lower, ES was "0." Observations showed ES as a strong predictive indicator, significantly correlating with mortality, Ranson criteria, and CT findings. Among 38 patients with ES "0" or "1," mortality was 5%, while among 35 patients with ES "2," mortality was 29%.
Amylase and its isoenzymes: As mentioned, serum amylase levels do not predict pancreatitis prognosis or severity, and their diagnostic value is limited due to nonspecificity.
The clinical value of amylase isoenzymes lies in: on one hand, they can help identify diseases with non-pancreatic hyperamylasemia, such as mumps, cerebrovascular diseases, respiratory diseases, renal failure, gastrointestinal ulcer perforation, biliary tract diseases, liver cirrhosis, and post-major surgery, where elevated amylase levels may occur while the P-type (pancreatic type) isoenzyme remains normal. On the other hand, they are used for screening tests of late-stage [third-stage] complications. Some literature mentions that the pancreatic isoenzyme P3 is closely related to pseudopancreatic cysts, with an elevated P3/P2 ratio in the blood and cyst fluid of patients with this seasonal disease. It has been observed that in patients with acute pancreatitis, the presence of P3 and persistent trypsin elevation at discharge often indicates the onset of late-stage [third-stage] complications. It is also suggested that P3 is the most sensitive indicator for complications of acute pancreatitis.
There are two types of isoenzymes, one originating from the pancreas (abbreviated as P) and the other from saliva (S). The S-type, in addition to coming from the salivary glands, also originates from spontaneous sweating glands, mammary glands, bronchi, ovarian tumors, and the prostate, among other locations. The P-type amylase isoenzyme completely disappears after pancreatic resection. The P-type amylase isoenzymes include P1, P2, and P3. The S-type includes S1, S2, and S3. In normal individuals, P-type amylase isoenzymes account for 40%, while S-type accounts for 60%. Among the P-type, P1 constitutes 80–90% of the total (P-type), P2 accounts for 10–20%, and P3 accounts for 0–4%. In normal individuals, the activity of P-type amylase isoenzymes in urine is higher than that of the S-type. Analysis of pancreatic amylase isoenzymes provides a basis for diagnosing the disease cause of hyperamylasemia. According to statistics, it can correct errors in 20–40% of hyperamylasemia cases.
Amylase-creatinine renal clearance ratio (ACCR): The normal ACCR ratio is 3.8–5.3%. A ratio >5–6% suggests acute pancreatitis. Warshaw reported that in 42 cases, 93% showed an elevated ratio, while none of the 44 cases in the control group showed an increase. Therefore, ACCR holds significant diagnostic value for acute pancreatitis, though the reason for this remains unclear. Two possibilities exist: one is that pancreatic amylase molecules, partially degraded by proteolytic enzymes, are more easily lost through glomerular filtration; the other is that kinins or other vasoactive substances released into the bloodstream during acute pancreatitis increase glomerular permeability and inhibit amylase reabsorption in the renal tubules. Some have found that ACCR may also rise in conditions other than acute pancreatitis (e.g., chronic kidney failure, diabetic acidosis, burns, severe liver failure). Thus, it only holds specific diagnostic significance when these conditions are excluded. Warshaw believes the value of ACCR lies in both confirming acute pancreatitis and ruling out non-pancreatitis hyperamylasemia. The calculation method for ACCR is:
(Urine amylase / Serum amylase) × (Serum creatinine / Urine creatinine) × 100
CRP and SPLA: Bouchler reported that a C-reactive protein (CRP) level >100mg/L detected acute necrotizing pancreatitis with a positivity rate of 95%. He considered it the most accurate "serum factor" for detecting acute necrotizing pancreatitis. Poulakaainen examined the average CRP values on the first day of admission in 53 cases of acute pancreatitis: hemorrhagic necrotizing pancreatitis showed 280mg/L, while mild cases showed 45mg/L. The serum phospholipase A2 (SPLA2) values for hemorrhagic necrotizing pancreatitis were 40–60 nmol LFFA/ml·min, while mild cases were <10 nmol LFFA/ml·min, with even higher values in critically ill patients. A CRP >140mg/L showed 100% sensitivity for detecting severe pancreatitis. The sensitivity and specificity of SPLA2 for detecting severe pancreatitis varied significantly depending on the chosen cutoff value. At a cutoff of 11 nmol LFFA/ml·min, specificity was 100%, but sensitivity was only 42%. Combining CRP and SPLA2 testing provides a robust method for diagnosing acute severe pancreatitis. CRP testing is convenient, rapid, and suitable for clinical use, whereas current SPLA2 testing methods are complex and time-consuming, preventing routine clinical application. Further simplification is needed.
(2) Ribonuclease: RNAase is derived from pancreatic cells that have collapsed due to hypoxia. Experimental observations by Warshaw and Lee showed that among patients with normal RNase levels, only 4% developed pancreatic necrosis and abscess formation. However, among the 13 cases with elevated RNAase levels, 11 exhibited pancreatic necrosis and abscess formation, with the elevation occurring within the first few days of pancreatitis onset. Therefore, RNAase is considered a potential monitoring indicator for pancreatic necrosis and advanced-stage pancreatic complications. Testing revealed that this enzyme in the blood of patients with necrotizing pancreatitis was 10 times higher than normal. Recently, it has also been discovered that elevated RNAase levels are not specific to acute necrotizing pancreatitis and can also rise in other conditions such as pancreatic cancer, leukemia, extensive burns, trauma, and renal failure.
(3) α1-antitrypsin and α2-macroglobulin: α1-antitrypsin is an acute-phase reactant that rises rapidly during acute pancreatitis, while α2-macroglobulin levels decrease with increasing severity (grade III). The sensitivity of α1-antitrypsin in detecting pancreatic necrosis is 77%, and that of α2-macroglobulin is 85%.
In 1985, Beger analyzed dozens of laboratory and clinical parameters in 85 cases of acute necrotizing pancreatitis and identified four generation and transformation parameters, along with contrast-enhanced CT, as the basis for monitoring necrotizing pancreatitis, with an accuracy rate exceeding 80% (Table 1).
Table 1 Detection Rates in 85 Cases of Necrotizing Pancreatitis (Beger)
| Necrotizing Pancreatitis/Interstitial Edematous Pancreatitis | Detection Rate (%) |
| C-reactive protein >120 mg/L (NL <40 mg/L) | 93 |
| Contrast-enhanced CT showing pancreatic involvement >50% | 88 |
| Lactate dehydrogenase >270 IU/L (NL 60–200 IU/L) | 87 |
| α 1 -antitrypsin >4.5 g/L (NL 1.5–3 g/L) | 83 |
| α 2 -macroglobulin <1.3 g/L (NL 1.3–2.7 g/L) | 82 |
The table indicates that CRP and LDH have the highest detection rates. Their elevation not only indicates acute pancreatic inflammation but also suggests pancreatic necrosis, making them highly valuable for distinguishing whether acute pancreatitis involves necrosis.
(4) Apolipoprotein A 2 : Apolipoprotein A 2 (APO-AII) decreases significantly in acute pancreatitis. Schender et al. measured the levels of this substance in the serum of 20 acute pancreatitis patients. Except for one deceased patient with an APO-AII concentration of 21.6 mg/dL, the other five deceased patients had APO-AII levels below 20 mg/dL. The diagnostic accuracy of this method for fatal pancreatitis reached 80%. The mechanism behind the decrease in APO-AII remains unclear and requires further investigation.
(5) Serum lipase: This method is commonly used for diagnosing acute pancreatitis. In the past, due to the lengthy detection time for serum lipase (requiring 24 hours), it was difficult to meet emergency needs. Additionally, since its peak occurs 72–96 hours after onset, it was less frequently used. Nowadays, the method has been improved, simplified, and accelerated, with results available in just over 10 minutes. Sensitivity and specificity have also been enhanced. Heming Way used an immunoassay to measure lipase activity, achieving a sensitivity of 100% and specificity of 96%, with no false negatives. Another advantage is that this enzyme remains detectable in the blood for an extended period, allowing for prediction.
(6) Trypsinogen Activation Peptide: The trypsinogen activation peptide (TAP) in the urine of patients with acute pancreatitis is specifically measured by immunoassay to early predict the severity of acute pancreatitis. Based on Gudgeon's detection of TAP and clinical control results, the maximum sensitivity and specificity separation value is 2 nmol/L. At admission, the predictive sensitivity was 80%, specificity was 90%, and accuracy reached 87%. Among those with TAP ≥ 2 nmol/L, 75% had one or more severe complications. Among those with TAP < 2 nmol/L, 92% had no complications.
(7) Others: Berry found that the plasma fibrinogen level upon admission is a reliable prognostic factor for acute pancreatitis. When it exceeds 6.0 g/L, it indicates severe pancreatitis.
Immunoreactive trypsin (IRT) and elastase-1 can improve the sensitivity and specificity of acute pancreatitis diagnosis, but they cannot be used to determine severity grade III.
There are numerous laboratory diagnostic methods for acute pancreatitis, each with its limitations. Close integration with clinical findings is essential to enhance their diagnostic value. Further efforts are needed to explore how to improve their positive rate and specificity.
II. Imaging Examination
Imaging examinations for acute pancreatitis provide more reliable evidence for diagnosis and prognosis monitoring.
(1) Ultrasound Examination:
Acute edematous pancreatitis: The pancreas shows diffuse enlargement of varying degrees, with homogeneous hypoechoic (i.e., weak echo) parenchyma, appearing as sparse gray spots. The pancreatic margins are generally regular and clear, with surrounding vessels often clearly visible. Some patients (about 1/6) may show localized inflammatory masses on ultrasound, while the pancreatic duct is usually normal, with about 8% of patients exhibiting grade I dilation. Approximately 1/3 of acute pancreatitis patients show no abnormalities on ultrasound. In the remaining 2/3 of patients, no abnormalities may be detected by B-ultrasound within the first 12–24 hours of onset.
Acute hemorrhagic necrotizing pancreatitis (AHNP): The pancreas exhibits diffuse scattered hypoechoic areas interspersed with irregularly distributed medium to high echoes; irregularly shaped high-echo masses; in cases of severe hemorrhage, corresponding anechoic areas may appear in the hematoma region, with hypoechoic deep areas and floating signs. When the pancreas is significantly swollen (especially the head), it may compress the inferior vena cava and the superior mesenteric vein, causing indentation or flattening of the vascular anterior wall, presenting as parallel linear echoes. The common bile duct may show grade I to grade II dilation. The main pancreatic duct may be obstructed to varying degrees due to inflammation, edema, or spasm, with an increased diameter. In cases of peritoneal effusion, lateral decubitus positioning may reveal anechoic fluid areas. About 9% of patients may exhibit localized inflammatory pancreatic masses. Notably, 20–60% of patients cannot undergo ultrasound due to bowel gas, and ultrasound struggles to distinguish between fluid-filled abdominal masses and solid necrosis. Intraoperative ultrasound is helpful in determining pancreatic necrosis and guiding surgical drainage.
(2) CT Examination and Prognostic Evaluation:
Nordestgaard classified early pancreatitis on CT into four grades: Grade A, normal; Grade B, simple pancreatic enlargement; Grade C, inflammation involving one peripancreatic region; Grade D, inflammation involving two or more peripancreatic regions. Grades A and B patients show no mortality or complications, while 31% of Grade C patients have severe complications, and 44% of Grade D patients have critical complications.
CT findings of hemorrhagic necrotizing pancreatitis: The pancreas shows diffuse enlargement with blurred boundaries. High-density imaging (CT value >60 Hu) suggests hemorrhage.
Intra- or extra-pancreatic fluid collections: Commonly seen in the ventral lesser sac or left anterior pararenal space, with thickening of the renal fascia.
Large soft tissue masses within the pancreatic bed, containing necrosis or liquefaction, indicate phlegmon formation, which may develop into abscesses if secondary infection occurs.
In hemorrhagic necrotizing pancreatitis, extrapancreatic involvement may affect the lesser sac, root of the mesenteric vessels, left and right retrocolic regions, left and right renal regions, and the retroperitoneum. Therefore, CT scans should analyze changes in these regions. CT is highly valuable for diagnosing acute hemorrhagic pancreatitis and assessing prognosis. It achieves a diagnostic accuracy of over 90% for acute pancreatitis and enables early confirmation of pancreatic necrosis, which is clinically challenging to determine initially.
CT contrast-enhanced scan for pancreatitis: The changes in pancreatic density before and after enhancement are more significant for the pathological changes of pancreatitis. Without contrast agent (unenhanced), the pancreatic density shows a decreasing trend from head to tail. The average density decreases from 43Hu in the head to 36Hu in the tail. After contrast injection, the density of the main stirred pulse increases by 3 times, while the pancreas only increases by 2 times. In the absence of necrosis, there is no significant difference in the average contrast density of the pancreas or in the contrast density of the pancreatic head, body, and tail sections. The density of the pancreatic tail is >50Hu, whereas in cases with pancreatic necrosis, none exceed 50Hu, and the average pancreatic and segmental contrast enhancement, as well as the ratio of pancreatic to main stirred pulse density, are significantly reduced. This examination should not be performed if there is significant hypovolemia.
ERCP and selective stirred pulse contrast: It is not suitable for acute pancreatitis, especially hemorrhagic necrotizing pancreatitis, and may instead exacerbate pancreatic damage.
Acute pancreatitis is divided into two types: mild and severe. The former involves minor damage to the pancreas, characterized by simple edema with little exudation and almost no positive findings on CT scans. The latter, however, entails severe pancreatic damage, manifesting as extensive hemorrhage and necrosis. Acute edematous pancreatitis accounts for about 90% of cases, with relatively few fatalities. In contrast, hemorrhagic necrotizing pancreatitis has a high mortality rate, ranging from 20% to 50%. Over the years, extensive research has been conducted on acute hemorrhagic necrotizing pancreatitis, particularly focusing on early diagnosis and systemic physiological disturbances. Several indicators have been proposed to minimize the mortality rate.
Due to the varying degrees of pathological changes and pathophysiological alterations in hemorrhagic necrotizing pancreatitis, it is currently almost impossible to rely on a single indicator—whether clinical manifestations, laboratory results, or imaging findings—to establish a definitive diagnosis. Moreover, no single indicator can fully elucidate the severity of pathological changes or predict prognosis. Since Ranson introduced in 1974 a set of indicators to assess the severity, surgical indications, and/or prognosis of hemorrhagic necrotizing pancreatitis, many scholars have proposed their own criteria for diagnosing acute hemorrhagic necrotizing pancreatitis and evaluating its prognosis, each with its own representativeness. Below is a brief introduction to several commonly used clinical criteria:
1. **Ranson Criteria**: In 1974, Ranson proposed 11 indicators to predict the severity of acute pancreatitis (Table 1).
**Table 1 Ranson Criteria**
| **At admission:** |
| 1. Age > 55 years |
| 2. White blood cell count > 16 × 109/L |
| 3. Blood glucose > 11.2 mmol/L |
| 4. Serum LDH > 350 IU/L |
| 5. Serum GOT > 250 IU/L |
| **Within 48 hours of admission:** |
| 1. Hematocrit decrease > 10% |
| 2. BUN increase > 1.79 mmol/L |
| 3. Serum calcium < 2 mmol/L |
| 4. Arterial PO2 < 8 kPa |
| 5. Base deficit > 4 mmol/L |
| 6. Estimated fluid sequestration > 6000 mL |
This standard has been applied for 20 years and is still used in clinical practice. Among these 11 criteria, the more positive indicators there are, the more certain the severity of the lesion becomes, and the worse the prognosis. In a 6-year report (1980), Ranson summarized the pathology and clinical features of severe pancreatitis and proposed that patients meeting 1–2 of the 11 criteria are classified as mild cases and can be treated with palliative therapy, with a mortality rate of 0.9%. If patients meet 3 or more of the 11 criteria, they are classified as severe pancreatitis and should undergo surgical treatment, with a very high mortality rate of 50–60%. In 1978, Ranson reported a group of cases illustrating the relationship between prognostic indicators and mortality rates (Table 2). The table shows that the more indicators a patient meets, the higher the mortality rate. For 0–2 indicators, the mortality rate is 0.9%; for 3–4 indicators, it is 16%; for 5–6 indicators, it rises to 40%; and for 7–8 indicators, it reaches 100%.
Table 2 Relationship between Ranson Prognostic Signs and Complication Mortality Rates
| Cases | Number of Prognostic Signs | |||
| 0-2 | 3-4 | 5-6 | 7-8 | |
| Number of Cases | 347 | 67 | 30 | 6 |
| Death or Severe Illness (%) (ICU Stay >7 Days) | 13 (3.7) | 27 (40) | 28 (93) | 6 (100) |
| 3 (0.9) | 11 (16) | 12 (40) | 6 (100) | |
2. In 1983, Bank reported his clinical criteria for assessing the prognosis of pancreatitis. Its characteristic is that, based on Ranson's criteria, it emphasizes the damage to vital extrapancreatic organs. Bank's criteria are not only diagnostic for hemorrhagic necrotizing pancreatitis but also indications for surgery (Table 3).
Table 3 Bank's Clinical Criteria
| Heart | Shock, tachycardia >130/min, arrhythmia, abnormal ECG |
| Lung | Dyspnea, rales, PaO2 <7.98 kPa, ARDS |
| Kidney | Urine output <20 ml/h, rising BUN and/or creatinine |
| Metabolism | Ca++, pH, decreased or falling albumin |
| Hematology | Decreased hematocrit, DIC (increased fibrin split products, decreased platelets) |
| Neurology | Dysphoria, confusion, localized signs |
| Hemorrhagic Manifestations | Signs, abdominal paracentesis |
| Severe Abdominal Distension | Severe paralytic ileus and ascites++ |
Bank's evaluation method: mild is 0, if there are one or several symptoms in any organ, it is severe pancreatitis.
3. Imrie (1976) proposed prognostic indicators for pancreatitis, totaling 9 items. These are somewhat similar to Ranson's criteria. The damage to other systemic organs (or systems) is not addressed. The criteria are listed in the following table (Table 4).
Table 4 Imrie Clinical Criteria
| Within 48 hours of admission: |
| 1. Age >55 years |
| 2. WBC >15×109/L |
| 3. Blood glucose >10.08 mmol/L |
| 4. BUN >16.07 mmol/L |
| 5. PaO2 <2 kPa |
| 6. Serum calcium <2 mmol/L |
| 7. Serum albumin <30 g/L |
| 8. LDH >60 IU/L |
| 9. SGOT or SGPT >200 IU/L |
4. With the widespread use of imaging techniques (ultrasound, CT, etc.) and their high detection rate for pancreatic damage, the prognostic indicators proposed by the above researchers are considered insufficient. Therefore, the Japanese Ministry of Health, based on its own standards, combined with Ranson's criteria and referencing Bank's systemic symptom criteria, further incorporated findings from ultrasound and CT imaging to establish a new standard for diagnosing grade III pancreatitis and predicting its prognosis. See the following table (Table 5).
Table 5 Diagnostic Criteria and Prognostic Factors for Acute Pancreatitis Grade III
| A. Clinical Symptoms | B. Blood Tests | C. Imaging Findings | |
| ⑴ Shock | ⑴ BE ≤ -3 mmol/L | ⑵ Ca ≤ 1.88 mmol/L | ⑵ CT Grade IV, V IV: Pancreatic enlargement, heterogeneous parenchyma, inflammation extending beyond the pancreas, with peripancreatic fluid collection V: Pancreatic enlargement, heterogeneous parenchymal density, inflammation extending beyond or surrounding the pancreas Ultrasound: Refer to CT criteria |
| ⑴ Respiratory distress | ⑴ Ht ≤ 30% (after fluid resuscitation) | ⑵ FBS ≥ 11.2 mmol/L | |
| ⑴ Neurological symptoms | ⑴ BUN ≥ 14.3 mmol/L or Cr ≥ 176.8 μmol/L | ⑵PaO2≦8kPa | |
| ⑴Signs of severe infection | ⑵LDH≧11.69μmol﹒S-/L | ||
| ⑴Hemorrhagic tendency | ⑵TP≦60g/L | ||
| ⑵PT≧15 seconds | |||
| ⑵Platelets≦100×109/L | |||
Grade III: In clinical signs and blood tests (1), even one positive item indicates severity; in blood tests and imaging (2), two or more items qualify as Grade III.
Grade III determination time: Within 48 hours (after admission), with continuous monitoring thereafter.
Diagnostic criteria for clinical signs:
Shock: Systolic blood pressure below 10.7kPa or above 10.7kPa but with manifestations of shock.
Dyspnea: Dependence on mechanical ventilation.
Neurological symptoms: Central nervous system symptoms with impaired consciousness (only pain response).
Signs of severe infection: Leukocytosis, body temperature above 38°C, positive blood culture or internal toxin test, and confirmed intra-abdominal abscess. Hemorrhagic tendency: Confirmed gastrointestinal bleeding or intra-abdominal hemorrhage.
Grade II: General condition is relatively good, with no significant circulatory failure or major organ dysfunction.
No clinical signs (1) or any item in blood tests (1). One positive finding in blood tests or imaging (2) indicates Grade II acute pancreatitis.
Grade I: General condition is good.
No items in (1) or (2), with blood tests nearly normal, indicating Grade I acute pancreatitis.
5. Atlanta Criteria: In September 1992, the International Symposium on Acute Pancreatitis in Atlanta established the following clinical classification criteria for acute pancreatitis:
Acute pancreatitis: An acute inflammatory process of the pancreas involving various local tissues or distant organ systems. Sudden onset, upper abdominal pain and varying degrees of abdominal signs, vomiting, fever, tachycardia, leukocytosis, elevated blood and urine amylase. Gross appearance: Pancreatic and peripancreatic necrosis and hemorrhage. Microscopic: Interstitial edema and fat necrosis.
Severe acute pancreatitis: Acute pancreatitis with organ failure and/or local complications such as necrosis, abscess, or pseudocyst; Ranson criteria ≧3, APACHE1 ≧8; organ failure includes shock (systolic BP <12kPa), pulmonary insufficiency (PaO2≦8kPa), renal failure (creatinine >177μmol/L), gastrointestinal bleeding (>500ml/24h), DIC (platelets ≦10×109/L), fibrinogen <1.0g/L, fibrin degradation products ≧80μg/ml, severe metabolic disturbances (calcium <1.87mmol/L). Local complications include necrosis, abscess, or pseudocyst.
Grade I acute pancreatitis: Accompanied by Grade I organ dysfunction, without the clinical manifestations of severe acute pancreatitis, and responsive to appropriate fluid therapy. If no improvement within 48–72 hours, complications should be considered. Contrast-enhanced CT shows normal pancreatic parenchyma. Pathological changes are mainly edema, with occasional pancreatic parenchymal and peripancreatic fat necrosis.
P


