| disease | Arrhythmia |
| alias | Cardiac Arrhythmia |
The normal heart rhythm originates from the sinoatrial node, with a frequency of 60 to 100 beats per minute (in adults), and is relatively regular. The sinoatrial node impulse sequentially excites the atria and ventricles through the normal atrioventricular conduction system, with a constant conduction time (0.12 to 1.21 seconds in adults); the conduction time of the impulse through the bundle branches, their branches, and the Purkinje fibers to the ventricular myocardium is also constant (<0.10 seconds). Cardiac arrhythmia refers to any abnormality in the origin of the heart rhythm, heart rate and rhythm, or impulse conduction. Terms such as "rhythm disturbance" or "irregular heartbeat" emphasize abnormalities in rhythm, whereas cardiac arrhythmia encompasses both rhythm and rate abnormalities, making it more precise and appropriate.
bubble_chart Etiology
Arrhythmias can occur in various types of organic heart diseases, with coronary atherosclerotic heart disease (commonly known as coronary heart disease), cardiomyopathy, myocarditis, and rheumatic heart disease (commonly known as rheumatic heart disease) being the most common, especially in cases of heart failure or acute myocardial infarction. Arrhythmias are also not uncommon in otherwise healthy individuals or patients with autonomic nervous system dysfunction. Other causes include electrolyte or endocrine imbalances, anesthesia, hypothermia, thoracic or cardiac surgery, drug effects, and central nervous system diseases. In some cases, the cause remains unknown.
[Cardiac Anatomy and Physiology Related to Arrhythmias]
(1) The Cardiac Pacemaker-Conduction System The myocardium is mostly composed of ordinary myocardial fibers, with a small portion being specially differentiated myocardial fibers, which form the cardiac pacemaker-conduction system.
The cardiac pacemaker-conduction system includes the sinoatrial node, internodal tracts, atrioventricular node, atrioventricular bundle (bundle of His), left and right bundle branches and their subdivisions, as well as the Purkinje fiber network. The sinoatrial node is located at the entrance of the superior vena cava in the right atrium and serves as the pacemaker controlling the heart's normal activity. The atrioventricular node is situated at the base of the interatrial septum, below the fossa ovalis, between the tricuspid valve leaflet and the coronary sinus opening, extending forward to form the atrioventricular bundle. The atrioventricular bundle, also known as the bundle of His, has a proximal portion that is the main trunk or penetrating part, passing through the central fibrous body and running along the interventricular septum membrane until the muscular apex of the septum (the branching portion). It first gives off the left posterior bundle branch, then the left anterior bundle branch, and itself continues as the right bundle branch, forming a three-branch system. The penetrating part, as it passes through the central fibrous body, lies between the mitral and tricuspid valve rings, while the branching portion extends to the interventricular septum membrane, muscular portion, and adjacent areas of the main stirred pulse valve. The left posterior bundle branch is thick and short, fanning out early; the left anterior bundle branch and the right bundle branch are slender, branching later. Both bundle branches run beneath the endocardium toward the apex, branching and rebranching, with fine branches anastomosing to form a network called the Purkinje fiber network, which penetrates deeply into the ventricular myocardium (Figure 1).
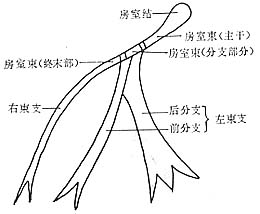
Figure 1 Schematic Anatomy of the Atrioventricular Conduction System
The sinoatrial node and the atrioventricular node are connected by three indistinctly bordered internodal tracts: anterior, middle, and posterior. The terminal portions of the internodal tracts that connect to the atrioventricular node, together with the atrioventricular node and the main trunk of the atrioventricular bundle, are collectively referred to as the atrioventricular junction (also known as the atrioventricular border or atrioventricular connection).
Between the atrial and ventricular myocardium lies a fibrous ring, preventing atrial excitation from being transmitted directly to the ventricles via the myocardium. The atrioventricular node and atrioventricular bundle are the sole normal pathway for atrioventricular conduction.
Blood supply to the cardiac conduction system: The sinoatrial node, atrioventricular node, and main trunk of the atrioventricular bundle are mostly supplied by the right coronary stirred pulse. The branching portion of the atrioventricular bundle, the left anterior bundle branch, and the right bundle branch receive blood supply from the left anterior descending branch of the left coronary stirred pulse, while the left posterior bundle branch is supplied by the left circumflex coronary stirred pulse and the right coronary stirred pulse.
The sinoatrial node and atrioventricular node are richly innervated by parasympathetic nerves, with the former receiving input from the right vagus nerve and the latter from the left vagus nerve.
(2) Electrophysiological Properties of Myocardium Myocardial cells exhibit automaticity, excitability, conductivity, and contractility, with the first three being closely related to arrhythmias.

The mechanism underlying automaticity is complex. It is currently believed that during the diastolic phase, autorhythmic cells experience an influx of sodium and/or calcium ions and an efflux of potassium ions across the membrane. When the influx of sodium and/or calcium ions exceeds the efflux of potassium ions, the negative membrane potential gradually decreases until the threshold potential is reached, triggering spontaneous depolarization and generating an action potential.
The automaticity of myocardial cells is influenced by the following factors: ① the maximum diastolic membrane potential; ② the threshold potential; ③ the slope of spontaneous depolarization. When the maximum diastolic membrane potential decreases, the depolarization slope becomes steeper, and the threshold potential approaches the resting membrane potential, the automaticity increases; conversely, the automaticity decreases. Among these three factors, the depolarization slope has the greatest influence (Figure 2). In a normal heart, the sinoatrial node has the highest automaticity. Before the spontaneous diastolic depolarization of other myocardial cells with automaticity reaches the threshold potential, they are already excited by the impulses transmitted from the sinoatrial node, and are referred to as the dominant pacemaker and latent pacemakers, respectively.

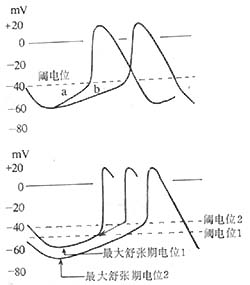
Figure 2 Factors Affecting Automaticity
Top: The slope of phase 4 depolarization changes from a→b, decreasing automaticity
Bottom: The threshold potential shifts from 1 to 2 (less negative), reducing automaticity. The maximum diastolic potential shifts from 1 to 2 (less negative), decreasing automaticity. The maximum diastolic potential shifts from 1 to 2 (more negative), while the threshold potential shifts from 1 to 2 (less negative), further reducing automaticity.
2. Excitability (or Irritability) When myocardial cells are stimulated by internal or external stimuli of appropriate intensity, they can depolarize and repolarize, generating action potentials. This property is called excitability or irritability. Stimuli insufficient to induce an action potential are termed subthreshold stimuli. The minimum stimulus intensity required to elicit an action potential is called the threshold stimulus. The excitability of myocardial cells is measured by the threshold stimulus intensity. If a stimulus must be stronger than the threshold to induce an action potential, it indicates decreased excitability of the myocardial cells. If a stimulus weaker than the threshold can induce an action potential, it indicates increased excitability of the myocardial cells.
Action Potential and Its Generation Mechanism: At rest, the interior of the myocardial cell membrane is negatively charged and relatively stable. This is due to the intracellular potassium ion concentration being 20–30 times higher than the extracellular concentration, causing potassium ions to flow out and carry positive charges with them. Meanwhile, larger anions that cannot easily pass through the cell membrane remain inside, preventing the outward movement of positively charged potassium ions. A threshold stimulus excites the myocardial cell, generating an action potential. First, the fast sodium channels on the cell membrane open. Since the extracellular sodium ion concentration is 10–20 times higher than the intracellular concentration and the membrane interior is more negative than the exterior, sodium ions rapidly flood into the cell, quickly shifting the membrane potential from negative to +30 to +40 mV, forming phase 0 of the action potential (depolarization). Subsequently, the sodium channels partially close, halting the rapid influx of sodium ions, while potassium ions flow out, causing the membrane potential to begin declining (phase 1, initial rapid repolarization). Next, calcium and sodium ions slowly flow in, and potassium ions slowly flow out, resulting in minimal changes to the membrane potential (phase 2, slow repolarization). Later, potassium ion outflow accelerates, rapidly reducing the membrane potential to the resting membrane potential level (phase 3, terminal rapid repolarization). The resting membrane potential during diastole is referred to as phase 4. In automatic cells, phase 4 involves sodium ion influx (in Purkinje cells) and/or reduced potassium ion outflow (in sinoatrial node cells), gradually reducing the membrane potential until the threshold potential is reached, triggering spontaneous depolarization. Non-automatic cells maintain a constant membrane potential during phase 4 (Figure 3). The time from the start of phase 0 to the end of phase 3 is called the action potential duration. In recent years, with advances in myocardial cell electrophysiology research and the application of voltage-clamp and patch-clamp techniques, new concepts have emerged regarding the ion channels and ion currents in the myocardial cell membrane.
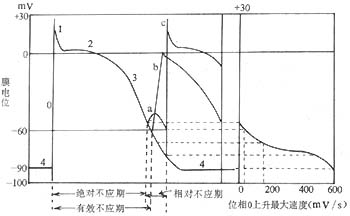
Figure 3 Left: Action potential and refractory period of myocardial cells Right: Membrane response curve a, b, c show the cell's response to stimuli during repolarization
a—Non-propagated local response b—First propagated response c—First normal response
The action potential curves of the sinoatrial node and atrioventricular node differ from those of other regions and exhibit the following characteristics: slow phase 0 depolarization, low amplitude, absence of phases 1 and 2, steep phase 4 depolarization slope, and low resting membrane potential and threshold potential (resting membrane potential -40 to -70 mV, threshold potential -30 to -40 mV, compared to -90 mV and -60 mV in ventricular muscle, etc.). The action potential duration is short (Figure 4). In recent years, it has been confirmed that the phase 0 depolarization in these two regions is caused by the slow influx of calcium and sodium ions, hence they are referred to as slow-response cells. In other myocardial cells, depolarization is caused by the rapid influx of sodium ions, making them fast-response cells. The electrophysiological properties of these two cell types are significantly different: slow-response cells have higher automaticity, poor conduction performance, and are prone to conduction block, whereas fast-response cells exhibit reliable conduction performance.
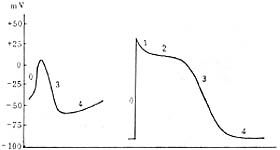
Figure 4 Left: Action potential of sinoatrial node cells (slow-response cells)
Right: Action potential of ventricular myocardial cells (fast-response cells)
The excitability of myocardial cells is influenced by the following factors:
(1) Membrane potential: When the membrane potential is below -55mV, no stimulus of any intensity can excite (or stimulate) the myocardial cells. Between -55mV and -80mV, stimuli stronger than the threshold can cause partial or complete depolarization of the cells. In the range of -55mV to -60mV, the partial depolarization of the cells cannot propagate to neighboring cells. Between -60mV and -80mV, the excitation generated by cell depolarization can propagate, but compared to normal conditions, phase 0 depolarization is slower, the amplitude is lower, and the action potential duration is shorter, resulting in lower excitability and slower conduction velocity. After depolarization, the excitability of myocardial cells changes with the degree of repolarization. The period before the membrane potential recovers to -55mV is the absolute refractory period, the period before it recovers to -60mV is the effective refractory period, and the range of -55mV to -80mV is the relative refractory period (Figure 3). Before the start of the relative refractory period, there is a brief vulnerable period (or excitable period), during which external stimuli are prone to form reentry and ectopic rhythms.
The refractory period of slow-response cells can extend beyond the completion of repolarization. When the action potential duration is prolonged, the refractory period correspondingly lengthens. Slow heart rate, hypokalemia, and the effects of quinidine-like drugs prolong the action potential duration and thus extend the refractory period.
(2) Membrane responsiveness: The depolarization response of myocardial cells at different membrane potentials is called membrane responsiveness, which can be represented by the membrane responsiveness curve (Figure 3). At the same membrane potential, myocardial cells with faster phase 0 depolarization speed and higher amplitude have stronger membrane responsiveness, higher excitability, and their membrane responsiveness curve shifts to the left; conversely, they have weaker membrane responsiveness, lower excitability, and the membrane responsiveness curve shifts to the right.
(3) The difference between the resting membrane potential and the threshold potential: When the resting membrane potential of myocardial cells is close to the threshold potential, the excitability is high; conversely, the excitability is low.
3. Conductivity Myocardial cells have the property of propagating impulses to neighboring cells, known as conductivity. Factors affecting conduction include: ① The effectiveness of the propagated impulse (speed and amplitude of phase 0 depolarization of the action potential); ② The excitability of the myocardial cells receiving the impulse; ③ The physical properties of the myocardial fibers, such as resistance to impulse propagation, which is influenced by fiber diameter, fiber orientation and structural consistency, as well as the size and distribution of intercalated discs. If the impulse itself is highly effective, the excitability of the myocardial cells receiving the impulse is also high, or the myocardial fibers have a large diameter and consistent orientation and structure with low resistance at the intercalated discs, then the conduction velocity is fast; otherwise, conduction is slow. The phase 0 depolarization speed of atrioventricular node cells is slow, and the amplitude is low, with inconsistent orientation and structure of the myocardial fibers within the node, resulting in slow impulse conduction and a higher likelihood of conduction disturbances.
Various parts of the heart are innervated by postganglionic fibers of the vagus and sympathetic nerves. The vagus nerve is mainly distributed in the sinoatrial node, atria, atrioventricular node, and the proximal His bundle, releasing acetylcholine to slow the phase 4 depolarization rate, thereby reducing the automaticity of the sinoatrial node and causing depolarization of latent pacemakers. The vagus nerve also shortens the refractory period of atrial muscle and prolongs the refractory period of the atrioventricular node, leading to corresponding conduction abnormalities. The sympathetic nerves are distributed throughout the heart, with richer innervation in the sinoatrial and atrioventricular nodes, releasing norepinephrine to increase the automaticity of the sinoatrial node and ectopic pacemakers, shorten the refractory period, and accelerate impulse conduction. The right stellate ganglion primarily innervates the sinoatrial node and atria, while the left stellate ganglion mainly innervates the ventricles.
Other factors that affect heart rate and rhythm, leading to arrhythmias, include hormones (adrenal cortical and medullary hormones, thyroid hormones, pituitary hormones, etc.), electrolytes (primarily potassium, sodium, calcium, magnesium), changes in blood pH, as well as oxygen and carbon dioxide tension.
[Mechanisms of Arrhythmia]
Results from extensive studies on single cells, isolated muscle strips, and animal cardiac electrophysiology indicate that arrhythmias can arise through various mechanisms, such as reentry, altered automaticity, triggered activity (caused by afterdepolarizations), and modulated parasystole. However, due to technical limitations, current in vivo human cardiac research is primarily limited to reentry mechanisms. Clinical examinations cannot yet determine the electrophysiological mechanisms of most arrhythmias, let alone distinguish their underlying ionic current mechanisms.
Table 1 Causes of Arrhythmia
| Abnormal Impulse Formation | Abnormal Impulse Conduction | Combined Abnormal Impulse Formation and Conduction |
| 1. Abnormal Automaticity Normal Automaticity | 1. Conduction Delay and Block (Sinoatrial Block, Atrioventricular Block, etc.) | 1. Parasystole 2. Phase 4 Depolarization Leading to Conduction Delay |
| Abnormal Automaticity | 2. Unidirectional Block and Reentry | |
| 2. Triggered Activity Early Afterdepolarization | 3. Conduction Block, Electrotonic Conduction, and Reflection |
The electrophysiological mechanisms of arrhythmias primarily include abnormal impulse formation, abnormal impulse conduction, or a combination of both (Table 1). Abnormal impulse formation occurs in: ① Normal automaticity, where the normal pacemaker (dominant or latent pacemaker) exhibits phase 4 depolarization that is either too rapid or too slow. ② Abnormal automaticity, where normally non-automatic fast-response cells (ventricular and atrial myocytes) and normally automatic fast-response cells (Purkinje fibers) develop abnormal automaticity when their membrane potential decreases to -50 to -60 mV due to pathological changes. The former transitions from non-automatic to automatic, while the latter exhibits enhanced automaticity (Figure 5). ③ Triggered activity following an action potential due to afterdepolarizations (Figures 6, 7).
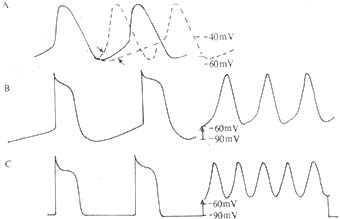
Figure 5 Mechanisms of Arrhythmia – Abnormal Impulse Formation
I. Normal Automaticity A: Phase 4 depolarization of the sinoatrial node, accelerated or slowed B (left): Phase 4 depolarization of Purkinje fibers
II. Abnormal Automaticity B (right): Purkinje fiber membrane potential drops to -60 mV, increasing automaticity C (left): Normal atrial or ventricular myocytes lack automaticity C (right): When membrane potential drops to -60 mV, abnormal automaticity emerges

Figure 6 Mechanism of Arrhythmia - Abnormal Impulse Generation Caused by Afterdepolarization Triggering
Programmed stimulation (.) and subsequent triggered excitation-induced spontaneous depolarization. Note that the afterdepolarization waves gradually increase to reach the threshold potential, leading to sustained tachyarrhythmia. Eventually, the afterdepolarization falls below the threshold potential, terminating the tachyarrhythmia.
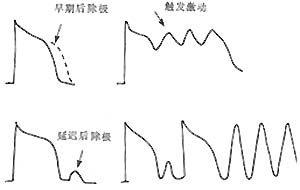
Figure 7 Early afterdepolarization (EAD) and delayed afterdepolarization (DAD) triggering abnormal impulse initiation (triggered activity).
Clinically, arrhythmias caused by abnormal impulse initiation under normal automaticity include sinus arrhythmia and escape rhythms. Abnormal automaticity can lead to accelerated atrioventricular junctional or ventricular rhythms, parasystole, or atrial or ventricular tachyarrhythmias. Afterdepolarizations are subthreshold depolarizations that occur during or after the repolarization phase of a preceding action potential, termed early afterdepolarization (EAD) and delayed afterdepolarization (DAD), respectively. When afterdepolarizations reach the depolarization threshold of slow-response cells, they can trigger one or multiple depolarizations. EAD occurs when myocardial cell repolarization is significantly prolonged due to various causes, such as increased extracellular potassium concentration, the effects of procainamide or high concentrations of catecholamines, or mechanical or stretch injury to Purkinje fibers. DAD, on the other hand, is associated with digitalis toxicity or other conditions that increase intracellular calcium levels. Programmed stimulation can induce and terminate tachyarrhythmias caused by afterdepolarization-triggered activity. Clinical electrophysiology cannot definitively distinguish between reentrant and triggered mechanisms of arrhythmias. Triggered activity may cause atrial or ventricular tachyarrhythmias, though its clinical significance and incidence remain under investigation.
Abnormal impulse conduction is often caused by changes in myocardial cell membrane properties, such as reduced action potential amplitude and upstroke velocity, decreased excitability, or reduced intercellular coupling, leading to slowed or blocked impulse conduction. This can result in the escape of latent pacemakers or reentrant tachyarrhythmias. The basic conditions for reentry formation are: ① Unidirectional block at one or more sites in the heart; ② Slow conduction along an alternative pathway; ③ Delayed excitation of myocardium distal to the block; ④ Re-excitation of myocardium proximal to the block. Nonuniform conduction inhibition in the heart creates significant electrophysiological differences between adjacent myocardial regions, forming two distinct conduction pathways with common proximal and distal channels. Under suitable conditions, an impulse travels from the proximal common channel along one pathway to the distal common channel, then re-enters the proximal channel via the other pathway, creating single or multiple reentrant circuits. Reentry can occur around fixed anatomical or electrophysiological conduction barriers when unidirectional block and slowed conduction are present. It may also manifest as a conduction vortex around a non-excitable central zone (microreentry—leading circle theory) or as electrotonic current reverse transmission across non-excitable myocardial gaps (reflection). (Figures 8, 9, 10).
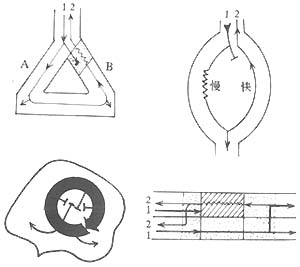
Figure 8 Mechanism of arrhythmia—unidirectional block and reentry.
Top left: Reentry between Purkinje fiber branches and ventricular muscle.
Bottom: Local reentry formed by electrophysiological dissociation within ventricular muscle.
Top right: Reentry due to longitudinal electrophysiological dissociation in the atrioventricular node.
Bottom: Reentry caused by electrophysiological dissociation between adjacent fibers in a myocardial bundle.

Figure 9 Reflection—reentry caused by electrotonic current transmission.
Top: Transmembrane potentials recorded by electrodes placed proximal and distal to a non-excitable segment of Purkinje fibers.
Lower right: The proximal impulse is blocked in the refractory segment, but the resulting electrotonic current allows the impulse to slowly conduct to the distal end.
Lower middle: Proximal impulse is blocked in the refractory segment, and the resulting electrotonic current is insufficient to cause distal excitation.
Lower left: The proximal impulse is delayed in conduction to the distal end via electrotonic current, then transmitted back to the proximal end, generating a second action potential—reflection.
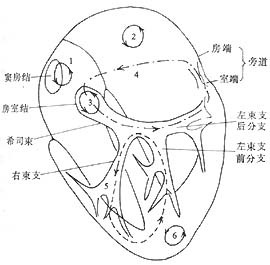
Figure 10: Mechanisms of Arrhythmia—Possible Sites of Reentry Circuits
1. Right atrium—sinoatrial node-atrial junction; 2. Left atrium; 3. Within the atrioventricular (AV) node; 4. AV reentry via accessory pathways; 5. Bundle branch reentry via the His-Purkinje system; 6. Within the ventricular myocardium.
Clinically, there is substantial evidence suggesting that the mechanisms of AV nodal tachycardia and supraventricular tachycardia caused by accessory pathways are reentry, with the reentry pathways being dual pathways within the AV node and the reentry loop involving the atrium, AV node, His-Purkinje system, ventricle, and retrograde conduction via the accessory pathway back to the atrium. Many supraventricular and ventricular tachyarrhythmias are due to microreentry within the atria or ventricles (Table 2).
When abnormal impulse formation coexists with abnormal impulse conduction, an ectopic pacemaker may be protected by surrounding entrance block, preventing invasion by neighboring excitation waves and maintaining its own depolarization rhythm, resulting in parasystolic rhythm. The interaction between abnormal impulse formation and conduction can alter the degree of entrance or exit block, leading to acceleration, deceleration, entrainment, or complete suppression of abnormal impulses, clinically manifesting as arrhythmias of varying rates. Recent reports also indicate that the rhythm of parasystolic impulses can be modulated by changes in sinus rhythm.
Table 2: Possible Mechanisms of Various Clinical Arrhythmias
| Mechanism | Clinical Arrhythmia |
| Reentry | AV nodal reentrant tachycardia, AV reentrant tachycardia via accessory pathways Sustained monomorphic ventricular tachycardia, bundle branch reentrant ventricular tachycardia, intra-atrial reentrant tachycardia |
| Abnormal Automaticity | Multifocal atrial tachycardia Certain types of atrial and ventricular tachycardia |
| Triggered Activity Due to Afterdepolarizations | Digitalis toxicity-induced atrial, AV junctional, and ventricular rhythms and tachycardias Accelerated junctional and ventricular escape rhythms Certain types of ventricular tachycardia |
[Classification of Arrhythmias]
Arrhythmias can be classified based on their mechanism of occurrence, the heart rate during the arrhythmia, and the severity of circulatory impairment and prognosis during the arrhythmia.
According to the mechanism of occurrence, arrhythmias are divided into abnormal impulse generation, abnormal conduction, and combined abnormalities of impulse generation and conduction. This classification method is primarily based on experimental research results and has limited practical value in clinical diagnosis, as current techniques are often unable to determine the electrophysiological mechanisms of arrhythmias. Additionally, the mechanisms initiating and sustaining certain rapid arrhythmias may differ. For example, ventricular premature beats caused by abnormal automaticity may lead to sustained ventricular tachycardia through a reentry mechanism.
Based on heart rate during the arrhythmia, arrhythmias can be classified as tachyarrhythmias (fast) and bradyarrhythmias (slow). In recent years, some scholars have proposed classifying arrhythmias into three categories—lethal, potentially lethal, and benign—based on the severity of circulatory impairment and prognosis. These two classification methods are simple and practical, combining clinical relevance to aid in the diagnosis, prevention, and treatment of arrhythmias.
The definitive diagnosis of arrhythmias mostly relies on electrocardiography (ECG), but a significant portion of patients can receive a preliminary diagnosis based on medical history and signs. Detailed inquiries about the heart rate and rhythm (regularity, palpitations, etc.) during episodes, as well as the onset, termination, and duration of episodes, are crucial. Symptoms such as hypotension, syncope or near-syncope, spasms, colicky chest pain, or heart failure during episodes, along with past triggers, frequency, and treatment history, help determine the nature of the arrhythmia.
Physical examination during an episode should focus on assessing the nature of the arrhythmia and its impact on hemodynamic status. Auscultation of heart sounds to evaluate the rate and regularity of ventricular contractions, combined with observations of atrial activity reflected in jugular venous pulsations, aids in the preliminary differential diagnosis of arrhythmias. A slow (<60 beats/min) and regular heart rate is commonly seen in sinus bradycardia, 2:1 or 3:1 atrioventricular (AV) block, complete AV block, sinoatrial block, or junctional rhythm. A fast (>100 beats/min) and regular heart rate is often due to sinus tachycardia, supraventricular tachycardia, atrial flutter, or atrial tachycardia with 2:1 AV conduction, or ventricular tachycardia. Sinus tachycardia rarely exceeds 160 beats/min, while atrial flutter with 2:1 AV conduction typically results in a fixed ventricular rate of around 150 beats/min. Irregular rhythms are most commonly caused by premature beats. Fast and irregular rhythms are often due to atrial fibrillation or flutter, or atrial tachycardia with irregular AV block. Slow and irregular rhythms are frequently seen in atrial fibrillation (after digitalis treatment), sinus bradycardia with sinus arrhythmia, or sinus rhythm with irregular sinoatrial or AV block. A regular rhythm with varying intensity of the first heart sound (cannon sound), especially accompanied by intermittent irregular enhancement of jugular venous pulsations (cannon waves), suggests AV dissociation, commonly seen in complete AV block or ventricular tachycardia.
The effect of carotid sinus massage on tachyarrhythmias can aid in differential diagnosis. To avoid complications such as hypotension or cardiac arrest, the procedure should be performed with the patient in a supine position under ECG monitoring. It should be used cautiously in elderly patients and is contraindicated in those with cerebrovascular disease. Massaging one carotid sinus at a time for no more than 5 seconds can halve the ventricular rate in atrial flutter or immediately restore sinus rhythm in supraventricular tachycardia.
The electrocardiogram (ECG) recording during an arrhythmia episode is a crucial basis for diagnosing arrhythmias. It should include a prolonged recording of lead II or V1. Pay attention to the morphology of P and QRS waves, the P-QRS relationship, and the PP, PR, and RR intervals to determine whether the underlying rhythm is sinus or ectopic. When there is independent atrioventricular activity, identify the origin of P waves and QRS complexes (select leads II, aVF, aVR, V1, and V5, V6). If P waves are not clearly visible, try increasing the voltage or paper speed to obtain a longer recording from leads where P waves are more prominent. If necessary, an esophageal lead or right atrial electrogram can be used to reveal P waves. If P waves are still not found despite consciously searching within the QRS, ST, and T waves using the above methods, consider the possibilities of atrial fibrillation, atrial flutter, atrioventricular junctional rhythm, or atrial standstill. By analyzing the nature and origin of premature or delayed beats one by one, the type of arrhythmia can ultimately be determined.
During the interictal period, physical examination should focus on identifying evidence of organic heart diseases such as hypertension, coronary heart disease, valvular disease, cardiomyopathy, myocarditis, etc. Non-invasive and invasive tests, including routine electrocardiogram (ECG), echocardiography, exercise stress testing, radionuclide imaging, and cardiac angiography, can help confirm or rule out organic heart disease.
Ambulatory ECG monitoring, through 24-hour continuous ECG recording, may capture episodes of arrhythmia, assess the influence of the autonomic nervous system on spontaneous arrhythmias, correlate symptoms with arrhythmias, and evaluate treatment efficacy. However, it may fail to detect infrequently occurring arrhythmias.
Invasive electrophysiological studies can not only diagnose the nature of bradyarrhythmias and tachyarrhythmias but also evaluate sinoatrial node and atrioventricular conduction system function during the arrhythmia-free interval using programmed electrical stimulation. These studies can induce supraventricular and ventricular tachyarrhythmias, localize the origin of arrhythmias, assess the efficacy of drug and non-drug therapies, and provide essential information for surgical, pacemaker, or ablation treatments.
Signal-averaged ECG (SAECG), also known as high-resolution body surface ECG, may detect ventricular late potentials on the body surface, which reflect delayed depolarization due to slowed ventricular conduction. The presence of ventricular late potentials provides a substrate for reentry, increasing the risk of ventricular tachycardia, ventricular fibrillation, and sudden cardiac death in patients with such findings.
Exercise testing may provoke arrhythmias during the interictal period, aiding in the diagnosis of intermittent arrhythmias. The induction of ventricular tachycardia during exercise testing after treatment with antiarrhythmic drugs (especially those that slow intraventricular conduction) may indicate a proarrhythmic effect of the medication.
bubble_chart Treatment Measures
The treatment of arrhythmia should include both acute management during episodes and prevention of recurrence. In addition to treating the {|###|}disease cause{|###|}, it can be divided into pharmacological and non-pharmacological approaches.
Treatment of the {|###|}disease cause{|###|} involves correcting cardiac pathological changes, adjusting abnormal pathophysiological functions (such as dynamic coronary stenosis, pump dysfunction, altered autonomic tone, etc.), and eliminating other triggers of arrhythmia episodes (such as electrolyte imbalances, adverse drug {|###|}side effects{|###|}, etc.).
Pharmacological treatment for bradyarrhythmias generally involves drugs that enhance myocardial automaticity and/or accelerate conduction, such as sympathomimetic agents (isoproterenol, etc.), vagolytic drugs (atropine), or alkalizing agents (molar sodium lactate or sodium bicarbonate). For tachyarrhythmias, drugs that slow conduction and prolong the refractory period are used, such as vagotonic agents (neostigmine, Rehmannia preparations), sympathomimetic drugs that indirectly stimulate the vagus nerve (methoxamine, phenylephrine), or antiarrhythmic drugs.
Currently, there are over 50 antiarrhythmic drugs in clinical use, often classified based on their effects on myocardial cell action potentials (Vaughan Williams classification) (Table 3). Class I drugs inhibit phase 0 depolarization and were once called {|###|}membrane{|###|} inhibitors. They are further subdivided into Ia, Ib, and Ic subclasses based on the degree of inhibition and their varying effects on refractory periods and conduction velocity, with quinidine, lidocaine, and encainide as representative drugs, respectively. Class II consists of adrenergic β-receptor blockers; Class III prolongs action potential duration and refractory periods, with amiodarone as a representative drug; Class IV comprises calcium channel blockers, with verapamil as a representative drug. In recent years, the Sicilian Gambit classification has also been introduced. Antiarrhythmic drug therapy does not eliminate the pathological tissue causing arrhythmias but modifies the electrophysiological properties of myocardial cells in the affected area, such as conduction velocity and/or refractory period duration. Long-term use of these drugs can cause varying degrees of adverse {|###|}side effects{|###|}, with severe cases potentially leading to fatal ventricular arrhythmias or heart block. Therefore, clinical application requires strict adherence to indications and familiarity with the properties of commonly used antiarrhythmic drugs, including half-life, absorption, metabolism, excretion, active metabolites, {|###|}dose{|###|}, and {|###|}side effects{|###|} (Tables 4, 5).
Table 3 Classification of Antiarrhythmic Drugs
| Class | Subclass | Electrophysiological Effects | Representative Agents | New Agents | ||
| Conduction Velocity | Refractory Period | AP Duration | ||||
| I (Sodium Channel Blockers)
| Ⅰa | ↓ | ↑ | Mostly↑ | Quinidine, Procainamide, Disopyramide | Ajmaline (ajmaline) |
| Pirmenol (pirmenol) | ||||||
| Aprindine (aprindine) | ||||||
| Ib | ↓ or ↑ | ↓ | ↓ | Lidocaine Phenytoin | Mexiletine (mexiletine) | |
| Tocainide (tocainide) | ||||||
| Ic | ↓ | ↑ | = | Encainide (encainide) | ||
| Flecainide (flecainide) | ||||||
| Lorcainide (lorcainide) | ||||||
| Propafenone (propafenone) | ||||||
| Ethmozin (ethmozin) | ||||||
| II (β-blockers) | ↓ | = | ↑ | Propranolol | Other β-blockers | |
| III (Prolonged action potential duration) | ↓ | =↑ | ↑ | Amiodarone | Sotalol (sotabl) | |
| IV Calcium channel blockers | ↓ | ↑ | Verapamil | Bepridil (bepridil) | ||
| Diltiazem | ||||||
Table 4 Common Usage of Antiarrhythmic Drugs for Rapid Arrhythmias
| Type | Drug Name | Indications | Dose and Usage | Main Adverse Reactions | ||
| Supra ventricular | Ventricular
| Therapeutic Dose | Maintenance Dose | |||
| Ⅰa | Quinidine quinidine | ++ | ++ | Oral 0.2~0.4g, every 2h, total 5 times/day, for cardioversion Oral 0.2g, 3~4 times/day, for premature beats | Oral 0.2~0.3g, 3~4 times/day, long-acting preparation 0.3g, every 8~12h | Hypotension, myocardial depression, intraventricular conduction block, severe ventricular arrhythmias, gastrointestinal reactions |
| Procainamide procainamide | ++ | ++ | IV 100mg every 5min, total 1.0~1.2g, IV infusion 0.5% at 5~10mg/min, total 1~2g Oral 0.5~1.0g, 5 times/day | Oral 0.25~0.5g, every 4~6h | Hypotension, intraventricular conduction block, ventricular arrhythmias, long-term use may cause lupus-like or wind-dampness-like symptoms | |
| N-acetylprocainamide N-acetylpro- cainemide | ++ | ++ | Oral 500~2500mg, every 6h | Oral 500~1000mg, every 6h | Same as above, does not cause lupus-like symptoms | |
| Disopyramide disopyramide | ++ | ++ | IV initial 100mg over 5~15min, then IV infusion 20~30mg/hour Oral 100~200mg, 4 times/day | Oral 100~200mg, every 6~8h | Cardiac conduction and myocardial depression, gastrointestinal reactions, dry mouth, urinary retention | |
| Antazoline antazoline | ++ | ++ | IV once 100~200mg Oral 0.1~0.2g, 4 times/day | Take 0.1~0.2g orally, 4 times a day | drowsiness, nausea, vomiting | |
| Pirmenol pirmenol | ++ | ++ | Intravenous administration of 2.5mg/kg, completed within 1 hour Oral 100~200mg, twice daily | Intravenous drip 0.25mg/min Oral 100~200mg, twice daily | Dry throat, headache, vertigo, insomnia, urinary retention, constipation, severe rapid ectopic arrhythmia | |
| Pyrozoline pyrozoline | ++ | ++ | Intravenous injection of 1.5mg/kg once Oral 0.2~0.3g, 3~4 times daily | Oral 0.2g, 2~3 times daily | Nausea, vomiting, skin discoloration, liver and kidney function damage, leukopenia | |
| Ajmaline ajmaline | ++ | ++ | Intravenous injection of 50mg (completed in 5~10 minutes) Oral 100mg, 3 times daily | Oral 50mg, 3 times daily | Drowsiness, vomiting, liver dysfunction, granulocytopenia, burning sensation with intravenous administration | |
| Ib | Lidocaine lidocaine | / | ++ | Intravenous injection of 50~100mg, 50mg every 5~10 minutes, total 250~300mg Intramuscular injection of 250~300mg | Intravenous drip 1~3mg per minute | Sinus arrest, atrioventricular block, myocardial contraction inhibition, drowsiness, speech and swallowing disorders, limb twitching |
| Phenytoin phenytoin | + | ++ | Intravenous injection of 100mg (completed in 5 minutes), then 100mg every 5~10 minutes, total 300~1000mg | Oral or intravenous injection of 0.1g, 3~4 times daily | Dizziness, drowsiness, granulocytopenia, local irritation with intravenous administration, hypotension, respiratory depression, sinus arrest, ventricular arrhythmia | |
| Mexiletine mexiletine | / | ++ | Intravenous injection of 100~200mg or intravenous drip of 250~500mg Oral 200~300mg, 3~4 times daily | Intravenous drip 1~2mg per minute Oral 200~300mg, 3~4 times daily | Bradycardia, hypotension, dizziness, nausea, vomiting | |
| Tocainide tocainide | / | ++ | IV drip 30~45mg per min, for 15min Oral 400~600mg, 3 times daily | Oral 400-600mg, 3 times/day | Vertigo, diplopia, gastrointestinal reactions | |
| Carbamazepine Carbamazepine | + | ++ | Oral 100-200mg, 3-4 times/day | Oral 100-200mg, 3 times/day | Vertigo, drowsiness, dyspepsia | |
| Aprindine Aprindine | + | ++ | IV infusion 200mg (2mg/min), then 100mg after 30min, and another 100mg after 6h Oral 50-75mg every 6h | Oral 25-50mg, 2-3 times/day | Dizziness, hand or finger tremor, ataxia, gastrointestinal reactions, granulocytopenia, myocardial depression | |
| Ic | Encainide Encainide | ++ | ++ | IV injection 1-2mg/kg over 15min Oral 25mg 3-4 times/day, may gradually increase to 50mg 3-4 times/day | Oral 25mg, 3-4 times/day | Dizziness, gastrointestinal reactions, rash, etc., severe arrhythmias |
| Flecainide Flecainide | ++ | ++ | IV injection 1-2mg/kg over 10min Oral 50-100mg twice daily, may gradually increase to 200mg twice daily | Oral 50-100mg twice daily | Dizziness, headache, nausea, weakness, nervousness, paresthesia, severe arrhythmias | |
| Lorcainide Lorcainide | ++ | ++ | IV infusion 2mg/min, or 100mg over 1h Oral 100mg twice daily, may gradually increase to 200mg twice daily | Oral 100mg twice daily | Insomnia, dreamfulness, anxiety, dizziness, headache, nausea, vomiting, paresthesia, severe arrhythmias | |
| Propafenone Propafenone | ++ | ++ | IV injection 70mg/dose over 3-5min Oral 150mg 3-4 times/day | Oral 300-600mg/day | Nausea, vomiting, headache, dizziness, orthostatic hypotension, AV and intraventricular conduction block | |
| Ethmozin Ethmozin | ++ | ++ | IV 1.8mg/kg, complete injection within 10min Oral 150~300mg, 3 times/d | Oral 100mg, 3 times/d | nausea, vomiting, headache, vertigo, ataxia, hypotension | |
| Cibenzoline cibenzoline | + | + | IV 1~2mg/kg Oral 30~80mg, 3~4 times/d, or 120~160mg, 2 times/d | Oral 30~80mg, 3 times/d, | nausea, vomiting, diarrhea, dry mouth, dizziness, lack of strength, drowsiness, liver function impairment, arrhythmia, cardiac function suppression | |
| Ⅱ | Propranolol propranolol | ++ | + | IV 0.5~1mg (complete injection in 5~10min) Oral 20mg, 3~4 times/d | Oral 10~20mg, 3 times/d | bradycardia, hypotension, heart failure, asthma, etc. |
| Pindolol pindolol | ++ | + | IV 0.2~1mg Oral 5~10mg, 3 times/d | Oral 5~10mg, 3 times/d | ||
| Atenolol atenolol | ++ | + | Oral 25~50mg, 1~2 times/d | Oral 25~50mg, 1 time/d | ||
| Metoprolol metoprolol | ++ | + | Oral 25~50mg, 2~3 times/d | Oral 25mg, 2 times/d | ||
| Betaxolol betaxolol | ++ | + | Oral 10~20mg, 1 time/d | Oral 10~20mg, 1 time/d | ||
| Acebutolol acebutolol | ++ | + | IV 10~20mg Oral 100mg, 3 times/d | Oral 100mg, 3 times/d | ||
| Esmolol exmolol | ++ | + | IV drip 25~300µg/kg per min | / | ||
| Flestolol flestolol | ++ | + | IV drip 0.5~10µg/kg per min | / | ||
| III | Bretylium bretylium | / | + | IV 250mg, every 6~8h Oral 0.1~0.4g, 3~4 times/d | Oral 0.1g, 3~4 times/d | Blood pressure fluctuations, nausea, vomiting, orthostatic hypotension |
| Amiodarone amiodarone | ++ | ++ | IV 250~500mg Oral 200mg, 3~4 times/d | Oral 200mg, 1~2 times/d | Bradycardia, skin discoloration, corneal microdeposits, thyroid dysfunction, severe arrhythmia, pulmonary fibrosis | |
| Sotalol sotalol | ++ | ++ | IV 20~60mg/dose, infused over 10min Oral initially 80~160mg, twice/d | Oral initially 80mg, twice/d | Adverse effects similar to propranolol, occasional neurological reactions and severe ventricular arrhythmias | |
| Bethanidine bethanidine | / | + | Oral initially 5~10mg, twice/d, then increase to 10~30mg, three times/d | Oral maximum maintenance dose 200mg/d | Same adverse effects as bretylium, mainly orthostatic hypotension | |
| IV | Verapamil verapamil | ++ | + | IV 5~10mg (infused over 5~10min). Oral 80mg, 3~4 times/d | Oral 80mg, 3~4 times/d | AV block, bradycardia, hypotension, heart failure |
| Bepridil bepridil | ++ | + | IV 3~4mg/kg once Oral 300~800mg, once/d | Oral 300~800mg, once/d | Adverse effects similar to verapamil, may cause ventricular arrhythmias | |
| Diltiazem diltiazem | ++ | / | IV 75~150µg/kg per dose Oral 60~90mg, three times/d | Oral 60mg, three times/d | Similar adverse reactions to verapamil, which may cause rashes | |
| Xin Ke Ding segontin | / | + | Oral 15~60mg, 3 times/day | Oral 15~30mg, 3 times/day | Bradycardia, hypotension | |
| V | Lanatoside C | ++ | + | IV 0.6~0.8mg, followed by 0.2~0.4mg after 2 hours | IV 0.4mg, once/day | Ventricular arrhythmia, atrial or atrioventricular junctional tachycardia, atrioventricular block, gastrointestinal reactions |
| Digoxin | ++ | + | IV 0.25~0.5mg, followed by 0.25mg after 4~6 hours Oral 0.25~0.75mg, 3 times/day for 2 days | Oral 0.2~0.5mg, once/day | ||
| Rehmannia glycosides | ++ | + | Oral 0.2~0.3mg, then 0.1mg every 6 hours, total 0.5~0.7mg within 1 day | Oral 0.05~0.1mg, once/day | ||
| Neofoside | ++ | + | Oral 0.1~0.2g (purified product 0.5~1mg), once/day | Oral 0.1~0.2g (purified product 0.5~1mg), once/day | ||
| Other
| Neostigmine | + | / | IM 0.5~1.0mg IV 5~10mg | / | Abdominal pain, nausea, muscle spasms, bradycardia |
| Edrophonium chloride | + | / | IV 5~10mg | / | ||
| Phenylephrine | ++ | / | IM 0.5~1.0mg to raise systolic blood pressure to 21.3kPa (160mmHg) | / | ||
| Methoxamine | ++ | / | IV 5~10mg to raise systolic blood pressure to 21.3kPa (160mmHg), then stop injection | / | Intraventricular conduction block, atrial arrest, gastrointestinal reactions | |
| Potassium chloride | + | + | Intravenous drip 0.3~0.5%, 1g per hour, total 1~2g. Oral 1~2g, 3~4 times/day | Oral 1g, 3~4 times/day | Intraventricular conduction block, atrial arrest, gastrointestinal reactions | |
| Magnesium sulfate | + | + | Intravenous injection 1~3g (dilute 10% 20ml injection by half), inject over 10 minutes, then intravenous drip. For magnesium deficiency: 6~9g on the first day, 2~3g on the second day | / | Hypotension, respiratory and cardiac arrest, bradycardia, atrioventricular and intraventricular conduction block, lack of strength, paralysis, drowsiness, unconsciousness | |
| Adenosine triphosphate | + | / | 5~10mg or 15~20mg diluted in 20ml normal saline, push within 5 seconds. Repeat after 3~5 minutes if ineffective | / | Atrioventricular conduction block, cardiac arrest, ventricular tachycardia | |
| Sophora | + | + | Intravenous injection 60mg. Intramuscular injection 20~40mg, oral 1.5~3.0g, 3 times/day | Oral 1.5~3.0g, 3 times/day | Nausea, acid reflux, upper abdominal pain | |
Table 5 Common Anti-Bradyarrhythmic Drugs Usage
| Drug Name | Indications | Dosage and Administration | Main Adverse Reactions | |
| Therapeutic Dose | Maintenance Dose | |||
| Isoproterenol | High-grade or complete atrioventricular block, sick sinus syndrome, cardiac arrest | Intravenous drip 1~3µg/min (1~2mg in 500ml 5% glucose solution, 1ml per minute) Sublingual 10~15mg every 3~4 hours | Same as above | Headache, vertigo, tremor, skin flushing, nausea, worsening of angina, rapid arrhythmia |
| Ephedrine | High-grade or complete atrioventricular block | Intramuscular or subcutaneous injection 15~30mg per dose Oral 25mg, 3 times/day | Oral 25mg, 3 times/day | Nervousness, vertigo, insomnia, tachycardia, hypertension |
| Adrenaline | High-grade or complete atrioventricular block, cardiac arrest | 0.1% 0.3~0.6ml intravenous, intramuscular, subcutaneous, or intracardiac injection; intravenous infusion 1~4µg/min | Intravenous infusion 1~4µg/min | Nervousness, pallor, tremor, hypertension, tachyarrhythmia |
| Atropine-like drugs | Sick sinus syndrome, atrioventricular block | Atropine 1mg subcutaneous, intramuscular, or intravenous injection; oral 0.3~0.6mg, 3 times/day Tangut anisodus alkaloid intravenous injection 10~20mg/dose, oral 5~10mg, 3 times/day Propantheline oral 15~30mg, 3 times/day | Oral 0.3~0.6mg, 3 times/day Oral 5~10mg, 3 times/day Oral 15~30mg, 3 times/day | Dry mouth, vertigo, skin flushing, urinary retention, aggravated glaucoma, tachyarrhythmia |
| Thyroid hormones | Sinus bradycardia or junctional rhythm, especially due to hypothyroidism | Thyroxine tablets oral 0.1~0.2mg/day Liothyronine sodium tablets 10~20ng/day | Same as above | Symptoms of hyperthyroidism, tachyarrhythmia |
| Adrenal corticosteroids | Atrioventricular block, especially due to inflammation | Intravenous infusion hydrocortisone 200~600mg (within 24 hours) Oral prednisone 10mg 4 times/day | Oral prednisone 10mg, 4 times/day | Manifestations of adrenal cortical hyperfunction, sodium retention, worsening diabetes, spread of inflammation, glaucoma, psychosis |
| Molar sodium lactate | Atrioventricular block or cardiac arrest caused by acidosis or hyperkalemia | Intravenous infusion rapid infusion of 25~50ml, followed by 5~7ml/kg, completed within several hours | Induced heart failure, alkalosis, hypokalemia, tachyarrhythmia | |
| Nicotinamide | Sick sinus syndrome | Intravenous infusion 400~800mg/day (may increase to 2000mg in some cases), for 2~4 weeks | Oral 2500mg/day | Nausea, vomiting, epigastric burning sensation, skin flushing, cutaneous pruritus |
Non-pharmacological treatments include mechanical methods to stimulate the vagus nerve, cardiac pacemakers, electrical cardioversion, electrical defibrillation, electrical ablation, radiofrequency ablation, cryoablation or laser ablation, and surgical therapy. Reflexive vagus nerve stimulation methods include eyeball compression, tuina of the carotid sinus, nose-pinching forced exhalation, and breath-holding. Cardiac pacemakers are primarily used to treat bradyarrhythmias, delivering low-energy electrical currents at a predetermined frequency to regularly stimulate the atria or ventricles and maintain cardiac activity. They are also employed to treat reentrant tachyarrhythmias and ventricular fibrillation by using programmed single or rapid sequential electrical stimuli to terminate reentry formation. Direct current cardioversion and defibrillation are used to terminate ectopic tachyarrhythmias and ventricular fibrillation, respectively. These methods involve applying high-voltage direct current briefly through the chest wall or directly to the heart, depolarizing both normal and abnormal pacemaker sites simultaneously to restore the sinoatrial node as the dominant pacemaker. To ensure safety, synchronized direct current cardioversion, which uses the patient's ECG R-wave to trigger discharge and avoids depolarization during the vulnerable period (thereby preventing ventricular fibrillation), is applied for converting atrial flutter, atrial fibrillation, and ventricular or supraventricular tachycardia. Non-synchronized direct current defibrillation is used for treating ventricular flutter and ventricular fibrillation. Electrical defibrillation and cardioversion are rapid, reliable, and safe, serving as the primary treatments for promptly terminating the aforementioned tachyarrhythmias, though they do not prevent recurrence.
In recent years, for severe and refractory ectopic tachyarrhythmias—such as recurrent sustained ventricular tachycardia with significant circulatory impairment, survivors of sudden cardiac death resuscitation, or patients with pre-excitation syndrome combined with supraventricular tachyarrhythmias exhibiting extremely rapid ventricular rates—it is advocated to first induce the arrhythmia via clinical electrophysiological testing with programmed stimulation. Subsequently, antiarrhythmic drugs are administered intravenously or orally, and the treatment plan is formulated based on the drug's efficacy in suppressing the induced arrhythmia. For patients unresponsive to drug therapy, the focus shifts to locating the reentrant pathway of the arrhythmia through clinical electrophysiology. Treatment options may then include transvenous catheter ablation (using methods such as electrocautery, radiofrequency, cryotherapy, laser, or selective alcohol injection into the coronary artery supplying the myocardial region of the reentrant pathway) or surgical intervention to disrupt the reentrant circuit.
The prognosis of arrhythmia is related to its cause, predisposing factors, progression, and whether it leads to severe hemodynamic disturbances. Arrhythmias occurring in individuals without underlying structural heart disease, such as premature beats, supraventricular tachycardia, and atrial fibrillation, generally have a favorable prognosis. However, in patients with long QT syndrome, ventricular premature beats may easily progress to polymorphic ventricular tachycardia or ventricular fibrillation, resulting in a poor prognosis. In patients with pre-excitation syndrome, atrial flutter or atrial fibrillation with a rapid ventricular rate not only tends to cause severe hemodynamic changes but also carries the risk of progressing to ventricular fibrillation. Nevertheless, most cases can be controlled with direct current cardioversion and medication, leading to a relatively good prognosis. Rapid ventricular arrhythmias and extremely slow heart rates, such as complete atrioventricular block, ventricular escape rhythm, and grade III sick sinus syndrome, can rapidly impair circulatory function and immediately threaten the patient's life. The prognosis of atrioventricular block caused by intranodal block differs significantly from that caused by bifascicular (trifascicular) block, with the former having a better prognosis and the latter a poor one. For arrhythmias occurring in the context of structural heart disease, if they do not cause significant hemodynamic disturbances and are unlikely to progress to severe arrhythmias, the prognosis is generally fair. However, if the underlying heart disease is severe, especially when accompanied by heart failure or acute myocardial ischemia, the prognosis is usually worse.


