| disease | Myasthenia Gravis (Surgery) |
Myasthenia gravis is an autoimmune disease affecting the neuromuscular junction, specifically the acetylcholine receptors on the postsynaptic membrane. It leads to skeletal muscle fatigue and weakness due to a reduced safety factor in junctional transmission. The clinical manifestations are diverse, involving not only striated muscle weakness but also visceral symptoms, which can result in loss of labor capacity and even death.
bubble_chart Pathogenesis
Since the proposal in 1960 that myasthenia gravis is an autoimmune disease, studies on α-bungarotoxin binding have revealed a reduction in the number of acetylcholine receptors, accompanied by a decrease in miniature potential amplitude and worsening of the condition. Unique anti-acetylcholine receptor antibodies have been detected in the sera of 90% of myasthenia gravis patients, drawing attention to the role of these antibodies in the {|###|}mechanism of disease{|###|} of myasthenia gravis. It is hypothesized that these antibodies reduce functional acetylcholine receptors, thereby impairing neuromuscular transmission. The mechanisms include: ① Accelerating the degradation of acetylcholine receptors on the postsynaptic {|###|}membrane{|###|}. ② Enhancing or accelerating the intracellular degradation of receptor-antibody complexes. ③ Reducing the synthesis of acetylcholine. ④ After binding to the receptor, occupying space and hindering the binding of acetylcholine to the receptor, thereby accelerating receptor degradation and destruction; selectively destroying the postsynaptic {|###|}membrane{|###|} with receptors through opsonization. Additionally, experimental studies have also demonstrated the role of these antibodies in the {|###|}mechanism of disease{|###|} of myasthenia gravis: ① A reduced humoral factor is present in the sera of myasthenia gravis patients, identified by sucrose gradient precipitation and ion-exchange chromatography as the immunoglobulin IgG, IgG3
. ② Injection of IgG from patient sera into experimental animals can reduce the amplitude of miniature endplate potentials and decrease acetylcholine receptors at the neuromuscular junction. ③ Injection of patient sera into rodents can passively transfer myasthenia gravis to these animals, and experimental allergic myasthenia gravis can also be passively transferred from one animal to another. These examples indicate that antibodies play a significant role in the destruction of receptors in the {|###|}mechanism of disease{|###|}. However, some studies have found that in patients after thymectomy, the antibody titers in their sera never turn negative; after plasma exchange, the reduction in antibody titers is transient, while clinical symptom relief can last for weeks or months. Therefore, it is believed that serum antibody titers are not directly correlated with clinical symptoms, possibly because antibody titer levels do not reflect the activity at the neuromuscular junction.Complement may also play a role in the {|###|}mechanism of disease{|###|} of myasthenia gravis. In 1977, Engel demonstrated the deposition of IgG and C3 complement on segments of the postsynaptic {|###|}membrane{|###|} where acetylcholine receptors are distributed and on fragments of degenerating junctional folds in the synaptic cleft. This suggests that after antibody binding to the receptor, complement is activated, damaging the postsynaptic {|###|}membrane{|###|} and leading to complement-mediated lysis of the postsynaptic {|###|}membrane{|###|}.
Recently, Sahashi used the immunoperoxidase method to demonstrate the presence of C9 on postsynaptic junctional folds and in the synaptic cleft, similar to the distribution areas of C3
complement. The activation of C9 complement causes irreversible {|###|}membrane{|###|} damage, supporting the role of complement-mediated lysis in the {|###|}mechanism of disease{|###|} of myasthenia gravis membrane destruction. This differs from other conditions, such as Duchenne muscular dystrophy, where junctional folds are destroyed without the involvement of IgG or Cq complement. Recent data suggest that the thymus may activate the alternative pathway of complement and accelerate this reaction process, and antibodies may also induce complement-mediated lysis of receptors.While the humoral immune mechanism is important, cell-mediated immune mechanisms cannot be excluded in the {|###|}mechanism of disease{|###|} of myasthenia gravis. Experimental studies have shown that in patients with late-stage [third-stage] onset, circulating T cells are reduced, primarily the T-cell subsets 3AI and OKT4. After thymectomy, these changes return to normal.
The immunological disease cause of myasthenia gravis remains inconclusive. Autoimmune diseases often occur on a genetic basis, with heredity potentially being the internal factor. Among external factors, many believe it is related to chronic sexually transmitted disease infections of the thymus. Patients with myasthenia gravis carrying HLA-A1, A8, B8, B12, or Dw3 are mostly female, with onset in youth, thymic hyperplasia, no tumors, low detection rates of acetylcholine receptor antibodies, ineffective response to anticholinesterase drugs, and good outcomes with early thymectomy. Patients with myasthenia gravis carrying HLA-A2 or A3 are mostly male, with onset after age 40, often accompanied by thymoma, and high detection rates of acetylcholine receptor antibodies. The above suggests that genetic factors play a significant role.
Thymic factors: At the beginning of this century, the thymus was considered unrelated to myasthenia gravis. However, data from thymectomies over the past 50 years indicate that thymic factors play a significant role in the {|###|}mechanism of disease{|###|}.Chronic, persistent {|###|}sexually transmitted disease{|###|} infections (thymic membrane inflammation) cause thymic epithelial cells to transform into myoid cells with new antigenic properties. These myoid cells closely resemble embryonic muscle cells and serve as precursors to mature lymphocytes. An increase in thymic B lymphocytes leads to abnormalities in B lymphocytes, resulting in the production of large quantities of autoantibodies. These myoid cells possess acetylcholine receptors whose antigenicity cross-reacts with those of striated muscle cells. Thus, the large number of antibodies produced against the new antigens of thymic myoid cells also target the acetylcholine receptors at the neuromuscular junctions of striated muscle cells, triggering the autoimmune disease—myasthenia gravis. A thymus affected by thymitis may also generate a population of cytotoxic T cells that damage neuromuscular junctions or produce a group of T helper cells that stimulate circulating lymphocytes to produce acetylcholine receptor antibodies.
The alleviation of symptoms after thymectomy may be due to the removal of the following sources: ① Acetylcholine antigens; ② Production of acetylcholine receptor antibodies; ③ Sensitized cytotoxic T cells directly attacking neuromuscular junctions; ④ Sensitized T helper cells promoting antibody production by peripheral lymphocytes; ⑤ Thymic factors activating the complement pathway, leading to complement-mediated lysis. However, thymectomy may be ineffective in some myasthenia gravis cases, possibly due to: ① Incomplete removal; ② Irreversible {|###|}injury{|###|} at the neuromuscular junction; ③ Lymphocyte populations in the spleen and peripheral lymph nodes still exerting thymus-like effects; ④ Long-lived peripheral T cells remaining active; ⑤ Heterogeneous {|###|}mechanism of disease{|###|}, with varying patient responses to thymic influences.
bubble_chart Clinical Manifestations
The incidence of myasthenia gravis in the general population is 1/20,000 to 1/75,000, and it can occur at any age, but it is more common in young women and elderly men. The first peak occurs at 20 years of age, and the second peak is around 50 years, with a male-to-female ratio of 1:2, while in young patients, this ratio reaches 1:4. Striated muscle weakness, fatigue, worsening during the day and improving in the evening, aggravation after activity, and relief after rest are the main symptoms of this disease. During myasthenic episodes, fluctuations can occur daily or even hourly. Muscle weakness may develop gradually or rapidly and can fully or partially recover. The initial symptoms are often simple extraocular muscle paralysis, but isolated limb, bulbar, or neck muscle weakness may also occur. About 56–60% of myasthenia gravis patients have extraocular muscle involvement. Eventually, 90% of patients exhibit ocular muscle weakness symptoms, such as ptosis, diplopia, and fluctuating ptosis during examination. Cogan's sign (ptosis of the levator palpebrae superioris after upward gaze) appears as the extraocular muscles are affected. The orbicularis oculi muscles also show weakness, and other cranial nerves may be involved, leading to potentially fatal complications such as dysphagia and respiratory distress. Patients in the late stage [third stage] often have injured masticatory muscles, inability to swallow, reliance on nasogastric feeding, inability to protrude the tongue, muscle atrophy, and an atypical three-grooved surface. Dysarthria, low voice, nasal speech, facial muscle weakness, a "sardonic smile" appearance, and weakness of neck flexors and extensors may force the patient to support their head with both hands. Over 80% of patients develop generalized myasthenia within one year of ocular muscle involvement.
Limb muscle weakness is mostly symmetrical, with proximal muscle groups more affected than distal ones, and the upper limbs more affected than the legs. Some patients may exhibit asymmetric weakness in individual muscles. Deep tendon reflexes are present but may temporarily disappear with repeated stimulation. Patients often report nonspecific sensations, but sensory examinations are normal. Autonomic nervous system changes manifest as pupillary abnormalities, bladder weakness, and profuse sweating, though these symptoms are uncommon. Occasionally, pyramidal tract signs may be observed, such as hyperreflexia in the limbs, which can elicit pathological reflexes. Mental stress can trigger sudden or gradual onset. After anesthesia or the use of muscle relaxants, myasthenia gravis presents as persistent muscle weakness.
The modified Osserman classification based on disease severity is as follows:
Type I: Only ocular muscle symptoms and signs, with no mortality.
II A Type: Grade I generalized myasthenia, slow onset, often involving ocular muscles and gradually affecting skeletal and bulbar muscles. No respiratory distress, poor response to medication. Limited activity, mortality.
II B : Grade II generalized myasthenia, involving bulbar muscles, preserved respiration, poor response to medication. Limited activity, low mortality.
Type III: Acute fulminant onset, early respiratory muscle involvement, severe bulbar and skeletal muscle impairment, highest incidence of thymoma. Limited activity, poor response to medication, but low mortality.
Type IV: Late stage [third stage] severe generalized myasthenia gravis. At least two years after the onset of Type I or II symptoms, progression may be gradual or sudden. Thymoma incidence ranks second. Poor response to medication and unfavorable prognosis.
The clinical symptoms of myasthenia gravis vary across age groups:
(1) Transient neonatal myasthenia gravis
Approximately 12–20% of newborns born to mothers with myasthenia gravis develop the condition, typically exhibiting signs at birth, though occasionally delayed by 12–18 hours. Common symptoms include difficulty sucking and swallowing, weak crying, and respiratory distress requiring assisted ventilation. Eyelid ptosis, facial muscle weakness, and poor facial expression are also observed. The primary cause of the disease is the transfer of maternal acetylcholine receptor antibodies across the placenta into the fetal bloodstream. As these antibodies degrade and are cleared from the fetal blood, clinical symptoms gradually improve, leading to the classification of this form as transient. Symptoms usually resolve spontaneously within 3 weeks, and tapering or discontinuing medication does not appear to carry a risk of recurrence. Critically ill infants should receive immediate treatment, including oral neostigmine (1–5 mg as needed) along with respiratory support and nutritional maintenance.
(II) Congenital Myasthenia Gravis
This type refers to newborns of normal mothers who suffer from myasthenia gravis, with a family history of the disease. 42% of cases manifest by age 2, and 66% before age 20. Affected infants lack acetylcholine receptor antibodies, and the disease mechanism is hereditary. There is structural deformity of the postsynaptic membrane; functional junctional folds are almost entirely absent, miniature structures are reduced, and acetylcholine receptors at the endplate are insufficient. Unlike transient neonatal myasthenia gravis, symptoms are persistent without complete remission. Symptoms often appear at birth or shortly after, with prominent involvement of extraocular muscles. Facial muscles are frequently affected, impairing feeding. Generalized muscle weakness is rare.
(III) Familial Infantile Myasthenia Gravis
This type involves infants of normal mothers with a family history of myasthenia gravis, such as siblings, and follows an autosomal recessive inheritance pattern. Severe respiratory and feeding difficulties are present at birth, distinguished from the previous two types by the characteristic feature of apnea. Respiratory failure often leads to infant death. Symptoms typically emerge before age 2, with a tendency for spontaneous remission and improvement with age. However, infections may trigger life-threatening apnea. Anticholinesterase drugs are effective, making early diagnosis crucial.
(IV) Cholinesterase Deficiency
This form of myasthenia gravis results from a lack of acetylcholinesterase in the subneural structures of the endplate, occurring in children and affecting eye muscles and muscles innervated by cranial nerves IX–XII. Trunk muscles are also involved, with proximal limb muscles more severely affected than distal ones. The Tensilon test is negative, and anticholinesterase drugs or guanidine (which increases acetylcholine release) are ineffective, whereas prednisone shows significant therapeutic effects.
(V) Juvenile Myasthenia
4% of all myasthenia cases manifest before age 10, and 24% before age 20, with a female predominance (4:1). Unlike the infantile form, genetic factors play a relatively minor role in this type, with immune mechanisms primarily driving the disease process. The course is slow and marked by fluctuations. Thymomas are rare.
(VI) Adult Myasthenia
In 70% of adult myasthenia cases, thymic hyperplasia is present, more common in younger individuals; 10–15% have thymomas, typically seen in older adults. Men progress faster than women, with lower remission rates and higher mortality. The clinical course features distinct exacerbations and remissions. In 3/4 of patients with ocular muscle involvement, symptoms generalize within 1–3 years, affecting pharyngeal muscles. In severe cases, multiple muscle groups may be asymmetrically involved. Most surviving patients enter a chronic, protracted phase with fewer episodes and milder symptoms.
(1) Drug Testing
Anticholinesterase drugs block the hydrolysis of acetylcholine at the synaptic cleft, prolonging its action and enhancing its ability to interact with acetylcholine receptors, thereby increasing miniature endplate potentials and improving the safety factor of neuromuscular transmission. These drugs can alleviate or reduce the clinical symptoms and electrophysiological abnormalities in patients with myasthenia gravis. The most commonly used anticholinesterase drug is edrophonium, which has a short duration of action and is effective in 95% of myasthenia gravis cases. A positive reaction confirms the diagnosis, while a negative reaction in individual cases does not rule out myasthenia gravis. It is recommended to perform this test in the evening or after exercise when muscle weakness is most severe. The ocular muscles are least sensitive to this drug, making diagnosis difficult in cases of myasthenia gravis limited to the ocular muscles.
Intravenous injection of 2–10 mg of edrophonium is administered, with an initial dose of 2 mg for sensitivity testing. For patients already taking anticholinesterase drugs, precautions should be taken to avoid exacerbating cholinergic muscle weakness symptoms. Allergic reactions and respiratory complications should be prepared for during the test. A positive reaction is typically evaluated using a triple-blind method, with normal saline and nicotinic acid as controls. Edrophonium can cause grade I headache and fever-like sensations, while nicotinic acid may replicate some symptoms but does not affect neuromuscular transmission, making it an ideal control.
If the reaction is transient and difficult to record with routine bedside techniques, long-acting anticholinesterase drugs with longer latency and duration of action may be used. Intramuscular injection of 1.5 mg of neostigmine can improve symptoms within 10–30 minutes, lasting up to 4 hours. If the reaction remains inconclusive, a long-term trial with oral anticholinesterase drugs for several weeks may be conducted.
(2) Electrophysiological Testing
The electrophysiological manifestation of myasthenia gravis is a reduction in the amplitude of miniature endplate potentials. The Jolly test involves repetitive stimulation of a nerve; normal individuals can tolerate stimulation at 40–50 times per second. In myasthenia gravis patients, stimulation at 2–3 times per second can cause an abnormal decrement in action potentials. Low-frequency repetitive electrical stimulation (2, 3, 5, 10, and 20 times per second) has been found to be diagnostically significant in domestic practice. The advantage of this test is its simplicity, but it is not highly sensitive, especially in the early stages of the disease, with approximately 50% of myasthenia gravis patients showing no changes.
A more sensitive method for assessing neuromuscular transmission is single-fiber electromyography. A single-fiber needle electrode is inserted between two muscle fibers innervated by the same motor nerve. Variations in the latency between two action potentials are represented as "jitter." In myasthenia gravis patients, neuromuscular transmission shows increased jitter or, in severe cases, blocking of one action potential with prolonged latency between the two. In 95% of myasthenia gravis patients with multiple muscle involvement, abnormal jitter is observed during testing, as jitter reflects the function of miniature endplate potential amplitude. This test can be used to monitor the clinical progression of myasthenia gravis. Its advantages include high sensitivity and early diagnostic capability, but it requires expensive equipment and neurophysiological expertise.
Stapedial reflex decay has also been used to diagnose myasthenia gravis, showing high sensitivity for ocular types but poor response in generalized cases.
(3) Serological Testing
The serum of myasthenia gravis patients contains many nonspecific antibodies, including anti-striated muscle, anti-nuclear, anti-thyroid, anti-gastric parietal, anti-sperm-producing, and anti-neuronal antibodies. Measuring their levels can provide diagnostic reference.
The special neurotoxin isolated and purified from cobra venom—α-bungarotoxin—has been found to irreversibly coagulate the active site of acetylcholine receptors in a targeted manner. This toxin can identify acetylcholine receptor antibodies and measure their quantity. The test involves applying the serum to be examined to acetylcholine receptor antigens obtained from human muscle, which have been embedded with 125I-labeled α-bungarotoxin to form a complex that coagulates at adjacent sites on the receptor. Subsequently, anti-human protein precipitates this complex. The level of acetylcholine receptor antibodies can be calculated based on the radioactivity of the precipitant (radioimmunoassay). Acetylcholine receptor antibodies in serum are highly specific for myasthenia gravis and can be detected in 90% of cases. They are only found when taking penicillamine or after snake venom inoculation, and acetylcholine receptors released from damaged muscles do not produce antibodies. It is generally believed that the level of these antibodies is unrelated to the patient's clinical symptoms, but patients with purely ocular symptoms tend to have lower antibody titers.
Recently, the enzyme-linked immunosorbent assay (ELISA) has been employed to measure antibodies, demonstrating higher sensitivity compared to radioimmunoassay. The coating used was α-Banded Krait toxin at 2.5 pmol/well, while the middle layer consisted of acetylcholine receptor extracted from electric eel electric organs at 0.2 pmol/well. The relative titer of the acetylcholine receptor was also measured, defined as the percentage of a patient's absolute titer at a given time phase relative to their highest absolute titer. Linear regression and correlation studies confirmed a close relationship between the severity of myasthenia gravis and the relative titer of acetylcholine receptor antibodies in the patient's serum. Additionally, serum immunoglobulin tests, including IgG, IgA, IgM, C3, and complement measurements, among other immunological assays, are all useful for diagnosing myasthenia gravis.
(IV) Chest X-ray Examination
Conventional chest radiography remains a relatively simple diagnostic method, achieving a diagnostic accuracy of 62% for thymoma. Mediastinal tomography of the thymic region can detect thymomas in 30% of cases with negative chest X-rays. Chest CT scans have a diagnostic concordance rate of 94%, capable of distinguishing between cystic and solid lesions, identifying calcifications, detecting smaller thymomas, and revealing signs of malignant thymoma, such as invasion of the pleura, lungs, or major blood vessels.
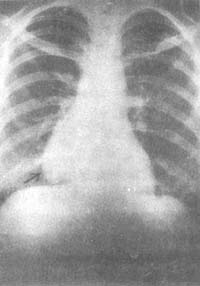
Figure 1 Posteroanterior chest radiograph shows a shadow adjacent to the right heart border.
Male, 58 years old, a patient with myasthenia gravis complicated by thymoma.

Figure 2 Right lateral chest tomogram shows:
An anterior mediastinal shadow (same case as above).
Myasthenia gravis can be associated with other diseases, such as rheumatoid arthritis, systemic lupus erythematosus, polymyositis, Sjogren's syndrome, ulcerative colitis, and other autoimmune diseases; it can also be associated with vitamin B12 deficiency, thyroid disorders, diabetes, parathyroid disorders, adrenal disorders, and vitiligo, which are considered part of the polyglandular failure syndrome. These associations may have genetic factors, as they are linked to histocompatibility antigens, particularly HLA-AI, A8, Dw3, which are risk factors for autoimmune diseases. In a given patient, a specific exposure may trigger an abnormal reaction. This inference is based on studies of monozygotic twins, where only one of the twins developed myasthenia gravis.
5% of myasthenia gravis patients have thyroid dysfunction. Sometimes, it is difficult to distinguish between the symptoms of myasthenia gravis and those of thyroid disorders, as both can cause proximal muscle weakness and ocular symptoms. Thyroid disorders are primarily endocrine conditions, whereas myasthenia gravis is more of an immune or genetic disorder. All thyroid disorders, including goiter, myxedema, Graves' disease, and Hashimoto's thyroiditis, can be associated with myasthenia.
1. Differential Diagnosis of Myasthenic Syndrome and Myasthenia Gravis
Myasthenic syndrome (Lambert-Eaton syndrome) is an acquired disease of the motor nerve terminals, caused by a reduction in the release of acetylcholine. The typical patients are males aged 50–70, complaining of weakness in the limb-girdle muscle groups, primarily the upper limbs, while the lower limbs, ocular muscles, or bulbar muscles are less affected or not involved. Deep tendon reflexes tend to be diminished or normal. This condition may be misdiagnosed as myasthenia gravis and is often associated with tumors, particularly small-cell lung cancer, with myasthenic symptoms appearing before tumor symptoms.
Myasthenic syndrome has an autoimmune basis, where pathogenic IgG antibodies cross-react with the presynaptic calcium ion system responsible for acetylcholine release. In affected nerve terminals, acetylcholine content and choline acetyltransferase activity are normal, indicating that acetylcholine synthesis and storage are intact. The defect arises from impaired vesicular release, reducing acetylcholine release. Due to decreased acetylcholine release at cholinergic autonomic regulatory sites, secondary familial dysautonomia may occur, manifesting as dry mouth, impaired ocular muscle function affecting accommodation at different distances, difficulty urinating, and constipation.
The typical electromyogram (EMG) in myasthenic syndrome shows a decremental response. Unlike myasthenia gravis, increasing exercise or high-frequency stimulation may paradoxically improve rather than worsen muscle weakness. Patients often exhibit a "sardonic grin" due to facial muscle weakness, yet muscle strength is relatively preserved. In contrast, myasthenia gravis patients show less pronounced facial changes but more severe muscle weakness. Skeletal muscle weakness in myasthenic syndrome responds poorly to anticholinesterase therapy, whereas 3,4-diaminopyridine, which enhances neurotransmitter release, is effective against neuromuscular and autoimmune neurological disorders. Drugs that increase acetylcholine release from presynaptic nerve terminals, such as calcium, may also be beneficial.
Myasthenia gravis patients are sensitive to non-depolarizing muscle relaxants but resistant to depolarizing muscle relaxants. In contrast, myasthenic syndrome patients are sensitive to both types of muscle relaxants.
When diagnosing this syndrome, bronchoscopy and mediastinoscopy should be performed. If lung cancer is suspected, thoracotomy may be necessary.
2. Differential Diagnosis of Other Diseases and Myasthenia Gravis Hysteria, thyroid disorders, and other neuromuscular diseases with myasthenic symptoms may sometimes be misdiagnosed as myasthenia gravis. However, the edrophonium chloride (Tensilon) test, single-fiber electromyography, and antibody level measurements can help differentiate these conditions.


