| disease | Hypercortisolism (Surgery) |
| alias | Cortisolism, Eisenmenger-Cushing Syndrome, Cushing's Syndrome, Cushing's Disease |
Cortisolism refers to the clinical syndrome caused by excessive cortisol in the body. In 1932, Cushing compiled 10 cases from the literature, combined with 2 cases he personally observed, and systematically described their clinical characteristics. Therefore, this condition was previously referred to as "Cushing's syndrome," and adrenal cortical hyperplasia caused by excessive secretion of adrenocorticotropic hormone (ACTH) by the pituitary gland was termed "Cushing's disease." Иценко (Itsenko) proposed in 1925 that this condition involved lesions in the pituitary gland and diencephalon, hence it was also called "Itsenko-Cushing syndrome." It is now certain that the direct cause of this group of conditions is excessive cortisol, so regardless of the underlying cause, it is uniformly referred to as "hypercortisolism," or simply "cortisolism."
bubble_chart Etiology
Cushing's syndrome can be classified into the following four types based on disease cause and pathological changes in the pituitary and adrenal glands (Figure 3):
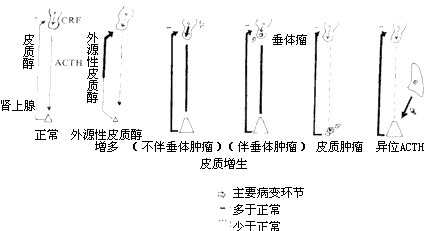
Figure 3 Pathogenesis of Cushing's Syndrome
(1) Iatrogenic Cushing's Syndrome Long-term, high-dose glucocorticoid therapy for certain diseases can lead to clinical manifestations of Cushing's syndrome, which is quite common in clinical practice. This is caused by exogenous hormones and can gradually resolve after discontinuation. However, prolonged high-dose glucocorticoid use can feedback-inhibit pituitary ACTH secretion, leading to adrenal cortex atrophy. Sudden withdrawal may result in a series of adrenal insufficiency symptoms or even crisis, so caution is warranted. Long-term ACTH use can also cause Cushing's syndrome.
(2) Pituitary Bilateral Adrenal Cortical Hyperplasia Bilateral adrenal cortical hyperplasia is caused by excessive ACTH secretion from the pituitary. The reasons include: ① Pituitary tumors, mostly basophilic adenomas, but chromophobe adenomas may also occur; ② No obvious pituitary tumor, but increased ACTH secretion, generally attributed to excessive secretion of corticotropin-releasing factor (CRF) by the hypothalamus. Clinically detectable pituitary tumors account for only about 10% of cases. In these cases, pituitary ACTH secretion has reached an abnormally high level, and elevated plasma cortisol is insufficient to trigger normal feedback inhibition, but high-dose dexamethasone can still suppress it.(3) Bilateral Adrenal Cortical Hyperplasia Caused by Extrapituitary Lesions Bronchial lung cancer (especially oat cell carcinoma), stony goiter (thyroid carcinoma), thymic carcinoma, nasopharyngeal carcinoma, and tumors originating from neural crest tissue may sometimes secrete an ACTH-like substance with similar biological effects, leading to bilateral adrenal cortical hyperplasia, hence termed ectopic ACTH syndrome. These patients often exhibit significant muscle atrophy and hypokalemia. The secretion of ACTH-like substances by the lesion is autonomous and not suppressed by high-dose dexamethasone. Symptoms gradually resolve after lesion removal or cure.
(4) Adrenal Cortical Tumors Most are benign adrenal cortical adenomas, with a minority being malignant adenocarcinomas. Tumor growth and adrenal corticosteroid secretion are autonomous and not controlled by ACTH. Due to excessive cortisol secretion by the tumor, pituitary secretion is feedback-inhibited, lowering plasma ACTH levels and causing significant atrophy of the non-tumorous adrenal cortex. In these patients, neither ACTH stimulation nor high-dose dexamethasone suppression alters cortisol secretion. Adrenal cortical tumors, especially malignant ones, often show markedly elevated urinary 17-ketosteroids.
Clinically, a few cases show nodular adrenal hyperplasia, an intermediate form between hyperplasia and adenoma. Plasma ACTH may be low, and high-dose dexamethasone shows no suppression.
Statistics indicate that clinically, 70% of cases are bilateral adrenal cortical hyperplasia due to pituitary lesions, benign adenomas account for 20–30%, malignant adrenal adenocarcinomas for 5–10%, and ectopic ACTH hypersecretion is very rare.
bubble_chart Clinical Manifestations
This disease is more common in women, with some cases occurring after {|###|}pregnancy{|###|}. The male-to-female incidence ratio is approximately 1:2. The age of onset is mostly between 15 and 40 years, but the youngest can be as young as 7 years old, and the oldest 62 years old. It is more common in adults than in children, and pediatric cases are often associated with cancer. If feminization occurs in males or masculinization in females, it often suggests the possibility of cancer. Adrenal cortical hyperplasia and adenoma progress relatively slowly, often taking 2–3 years after symptoms appear before medical consultation, whereas cancer develops rapidly and severely.
The clinical manifestations of this disease are caused by excessive cortisol, leading to metabolic disturbances of carbohydrates, proteins, fats, and electrolytes, as well as dysfunction of multiple organs.

(2) Plethora and striae: The skin becomes thin and atrophic, with thinning of the subcutaneous capillary walls, leading to facial redness and a plethoric appearance. Capillary fragility increases, making minor {|###|}injury{|###|} prone to bruising, especially on the upper arms, back of the hands, and inner thighs. Purple striae may appear on the abdomen, waist, armpits, thighs, and popliteal fossae, with an incidence rate of up to 3/4. These striae are generally wide and retain their color for a long time. They can occur not only in areas with abundant fat but also on the inner thighs and popliteal regions.
(3) Fatigue, weakness, and back pain: These are often the results of muscle atrophy and osteoporosis, particularly noticeable in the spine, pelvis, and ribs. Severe cases may lead to pathological {|###|}fracture{|###|}. Osteoporosis increases urinary calcium excretion and may sometimes complicate into kidney stones.
(4) Hypertension is relatively common and is associated with cortisol promoting angiotensinogen formation and mineralocorticoids causing water and sodium retention.
(5) Hirsutism, {|###|}alopecia areata{|###|}, and {|###|}acne{|###|}: Both men and women often exhibit excessive hair growth, which is particularly noticeable in women, sometimes even leading to beard growth. However, this is often accompanied by {|###|}alopecia areata{|###|}, possibly related to skin atrophy. {|###|}Acne{|###|} may occur on the face, chest, buttocks, and back.
(6) Sexual dysfunction: Patients often experience reduced libido. Men may develop {|###|}impotence{|###|}, while women may suffer from {|###|}amenorrhea{|###|}, menstrual irregularities, or reduced menstruation.
(7) Diabetes: Most cases are latent diabetes, manifested by elevated fasting blood glucose and diabetic curves in glucose tolerance tests, accounting for 60–90% of cases. A few cases present with clinical diabetes symptoms and glycosuria, termed steroid-induced diabetes. Patients often exhibit resistance to insulin therapy.
(8) Electrolyte metabolism and acid-base imbalance: Manifested as increased blood sodium and decreased blood potassium. Severe cases may develop hypokalemic, hypochloremic alkalosis. Patients may experience edema due to sodium retention.
(9) Reduced resistance to infection: Patients are prone to pyogenic bacterial, fungal, and certain viral infections. Once infected, the condition often fails to localize and spreads systemically, leading to severe sepsis and toxemia. Wound infections are slow to heal. Defensive reactions such as {|###|}fever{|###|} are suppressed, often resulting in misdiagnosis of {|###|}fistula disease{|###|} with serious consequences. If {|###|}acne{|###|} or tinea corporis is present on the trunk at the incision site, it may interfere with surgery.
(10) Other symptoms: Such as edema, liver dysfunction, exacerbation or bleeding of gastrointestinal {|###|}ulcer{|###|}, and mental disturbances.
bubble_chart Auxiliary Examination
The patient's hemoglobin and red blood cell count are slightly elevated, with a mild increase in white blood cells, a higher percentage of neutrophils, lower lymphocytes, and an eosinophil direct count of <50/mm3.
In some cases, blood sodium is elevated, while potassium and chloride are decreased, accompanied by alkalosis. Most cases show a diabetic curve in the glucose tolerance test, and some cases exhibit elevated fasting blood glucose or glycosuria. Plasma cortisol secretion exhibits a distinct diurnal variation: peaking in the early morning (10 ± 2.1 μg/dL) and gradually declining thereafter, with an average value of about 4.7 ± 1.9 μg/dL at 4 PM, reaching its lowest level before bedtime. If plasma cortisol concentration is measured every 4 hours and plotted on a graph to form a curve, it should appear V-shaped. However, in cases of hypercortisolism, the plasma concentration may exceed 30 μg/dL, and the V-shaped diurnal variation is lost.
Urinary 17-hydroxycorticosteroids exceed normal values (normal range: 5–15 mg/24h for males, 4–10 mg/24h for females). Urinary 17-ketosteroids may be normal or slightly elevated. If significantly elevated, especially >50 mg/24h, malignancy should be considered (normal range: 6–18 mg/24h for males, 4–13 mg/24h for females).
The diagnosis of Cushing's syndrome involves three aspects: confirming the disease diagnosis, determining the disease cause, and localization diagnosis.
(1) Confirming the disease diagnosis primarily relies on typical clinical symptoms and signs. These include central obesity, purple striae, hirsutism, sexual dysfunction, fatigue, etc. Additionally, a significant increase in urinary 17-hydroxycorticosteroid excretion, failure to suppress with a low-dose dexamethasone suppression test, and elevated blood 11-hydroxycorticosteroid levels with loss of diurnal rhythm confirm the diagnosis of Cushing's syndrome. Early or mild cases should be differentiated from simple obesity.
Low-dose dexamethasone test: Administering low-dose dexamethasone does not affect the measurement of urinary 17-hydroxycorticosteroids but can feedback-inhibit pituitary ACTH secretion. The method involves measuring 24-hour urinary 17-hydroxycorticosteroid excretion for six consecutive days. On days 3–4, oral dexamethasone 0.75mg is administered every 8 hours. The daily values are plotted on a graph and connected to form a curve. In normal individuals, urinary 17-hydroxycorticosteroid excretion decreases significantly two days after medication, with a reduction of more than half indicating significant suppression, which is normal. Conversely, if the reduction is insignificant or less than 50%, it indicates Cushing's syndrome.
(2) Disease cause diagnosis involves distinguishing between adrenal cortical adenoma, adenocarcinoma, pituitary tumor-induced cortical hyperplasia, non-pituitary tumors, or ectopic ACTH-secreting tumors causing cortical hyperplasia (Table 1).
Table 1: Methods for Differentiating the Causes of Cushing's Syndrome
| Adrenal Cortical Hyperplasia | Adrenal Cortical Adenoma | Adrenal Cortical Adenocarcinoma | Ectopic ACTH Syndrome | |
| Incidence | 60–70% | 29–30% | 1% | Rare |
| Gender | More common in females | More common in females | More common in females | More common in males |
| Disease Course | Long | Relatively long | Short | Short |
| Urinary 17-OH | ↑ | ↑ | ↑↑ | ↑↑ |
| Urinary 17-KS | ↑ | ↑ | ↑↑ | ↑↑ |
| ACTH Stimulation Test | ↑↑ | ↑ | - | - |
| Dexamethasone Suppression Test | ||||
| 2mg/d | - | - | - | - |
| 8mg/d | ↓ | - | - | - |
| Metyrapone Test | Positive response (no response in cases caused by pituitary adenoma) | No response | No response | No response |
| Vasopressin Test | Positive response | No response | No response | No response |
| Radionuclide Scanning | Bilateral concentration | Unilateral concentration | No imaging | No imaging |
| Other Imaging Diagnostic Methods | Normal adrenal glands, partial pituitary occupancy | Affected adrenal gland occupancy | Affected adrenal gland occupancy | Normal adrenal glands, possible detection of ectopic ACTH-secreting lesions |
1. X-ray Diagnosis of the Sella Turcica Pituitary tumors can compress the optic nerve, leading to temporal hemianopia. On X-ray spot films of the sella turcica, osteoporosis and decalcification of the sella floor and dorsum, absorption of the anterior and posterior clinoid processes, and enlargement of the sella turcica can be observed. Stratified films or CT scans can reveal smaller pituitary tumors. Secondary adrenal cortical hyperplasia caused by pituitary tumors accounts for about 10% of Cushing's syndrome cases.
2. ACTH Stimulation Test In cases of adrenal cortical hyperplasia, there is still a significant response to ACTH stimulation. The method is the same as the low-dose dexamethasone test, but on days 3-4, 20 units of ACTH are administered intravenously daily (added to 500-1000 ml of 5% glucose solution, infused over 8 hours). In adrenal cortical hyperplasia, the 24-hour urinary 17-hydroxycorticosteroid excretion increases by more than 50% compared to pre-injection levels, and the blood eosinophil count often decreases by 80-90% simultaneously. In adrenal cortical hyperplasia with small adenomas or nodular cortical hyperplasia, the response to the ACTH suppression test is similar to that of hyperplasia but may sometimes be weaker or less pronounced. In adrenal cortical tumors, because the normal adrenal cortex is in an atrophic state, there is no response or a very weak response. However, in cases with a short disease course, especially small adenomas or rapidly developing cortical carcinomas, the adrenal cortex outside the tumor has not yet atrophied, so there may still be a relatively significant response to this test. Ectopic ACTH-secreting tumors, due to the massive secretion of ACTH by the tumor, keep the adrenal cortex in a persistently hyperstimulated state, so there is no response to this test.
3. High-Dose Dexamethasone Suppression Test The method is the same as the low-dose dexamethasone test, but on days 3-4, 2 mg of dexamethasone is taken every 6 hours. A positive response is indicated by a reduction of more than 50% in the 24-hour urinary 17-hydroxycorticosteroid excretion compared to pre-medication levels. Ectopic ACTH-secreting tumors, cortical adenomas, and cortical adenocarcinomas have autonomous secretory functions and do not respond to this test. In contrast, cortical hyperplasia can show significant suppression and a positive response. In cases of cortical hyperplasia with small adenomas or nodular hyperplasia, although the ACTH stimulation test may be positive, high-dose dexamethasone cannot suppress their secretion (i.e., they can be stimulated but not suppressed). In such cases, other tests are needed to differentiate between cortical tumors and hyperplasia.
4. Metyrapone (Mepyrapone, Su4885) Test Metyrapone inhibits the 11β-hydroxylase enzyme, thereby blocking the conversion of 11-deoxycorticosterone to corticosterone and 11-deoxycortisol to cortisol. This leads to a decrease in plasma cortisol levels, which weakens the negative feedback inhibition and stimulates the pituitary gland to secrete large amounts of ACTH. Plasma ACTH levels rise (normal value at 8–10 AM: <100 pg/ml), and the synthesis of 11-deoxycortisol increases. Since 11-deoxycortisol is included in the measurement range of 17-hydroxy and 17-ketocorticosteroids, the urinary excretion of 17-hydroxy and 17-ketocorticosteroids also increases. Therefore, this test can assess the pituitary gland's reserve capacity to secrete ACTH. In cases of adrenal hyperplasia, the metyrapone test yields a positive response. However, in adrenal tumors, the tumor autonomously secretes large amounts of cortisol, suppressing the pituitary gland's ability to secrete ACTH, so the stimulatory effect of metyrapone is not observed. Similarly, in cortisol excess caused by pituitary adenomas, the pituitary gland autonomously secretes large amounts of ACTH, resulting in a negative response to the test.
5. Vasopressin Test Vasopressin has effects similar to CRF and can thus be used to assess the pituitary's reserve capacity for secreting ACTH. Patients with cortical hyperplasia show a positive response (increased blood ACTH and urinary 17-hydroxycorticosteroid excretion), while those with cortical tumors show a negative response. Vasopressin can cause coronary artery constriction, so it should not be used in elderly patients or those with coronary heart disease.
If all three tests—ACTH, metyrapone, and vasopressin—yield no response, adrenal cortical carcinoma may be suspected. If all three tests show positive responses, cortical hyperplasia is likely.
(3) Localization Diagnosis The primary goal is to locate adrenal cortical tumors to facilitate surgical removal. However, localization often also helps confirm the disease etiology.
1. Chest X-ray This can rule out lung cancer and pulmonary metastases.
2. Intravenous Pyelography (IVP) Assesses the condition of both kidneys. Larger adrenal tumors may displace the affected kidney downward.
3. Retroperitoneal Pneumography Due to the generally small size of adrenal cortical adenomas and the potential for misdiagnosis caused by massive retroperitoneal fat in obese individuals, this method is now rarely used.
4. Ultrasound Convenient and effective, with an accuracy rate exceeding 90% for locating adrenal cortical tumors.
5. CT Scan Can accurately localize most adrenal cortical adenomas larger than 0.5–1 cm in diameter.
6. Radioactive Iodine-Labeled Cholesterol Adrenal Scintigraphy Normal adrenal glands appear faint and symmetrical, with some individuals showing no imaging. In cortical hyperplasia, both adrenal glands show symmetrical but concentrated imaging. For cortical adenomas or carcinomas, the affected adrenal gland shows concentrated radioactivity, while the contralateral side does not appear. Some adenocarcinomas may show imaging, while others do not, possibly due to low hormone secretion per unit weight of tumor tissue, resulting in less cholesterol uptake and weaker radioactivity. This method is also useful for detecting residual adrenal tissue post-surgery, transplanted adrenal tissue, or ectopic adrenal tissue.
7. Adrenal Angiography Adrenal cortical tumors causing Cushing's syndrome are typically small and poorly vascularized, so adrenal angiography and venous blood sampling for cortisol measurement are not routinely performed. However, selective adrenal artery or venous angiography has been reported to visualize adrenal cortical adenomas. For tumors that cannot be localized by other methods, especially adrenal cortical carcinomas, adrenal angiography may be used for definitive diagnosis.
bubble_chart Treatment Measures
I. Surgical Therapy
1. Pituitary Tumor Resection Suitable for bilateral adrenal cortical hyperplasia caused by pituitary tumors, especially in cases with optic nerve compression symptoms. However, the tumor often cannot be completely removed, and it may affect other endocrine functions of the pituitary. If the resection is incomplete or the tumor cannot be removed, pituitary radiation therapy can be performed. If pituitary insufficiency occurs, necessary hormone supplementation should be provided. Bilateral adrenal cortical hyperplasia caused by pituitary microadenomas can be treated by selective resection of the pituitary microadenoma via the transsphenoidal approach using microsurgical techniques. This method is minimally invasive, does not affect pituitary function, and is a disease cause treatment, yielding good results. It has been widely adopted. If the microadenoma is not completely removed, symptoms may persist postoperatively; if the microadenoma is hypothalamic-dependent, recurrence may occur after surgery.
2. Adrenal Cortical Tumor Resection Suitable for adrenal cortical adenomas and adrenal cortical carcinomas. If the tumor can be accurately localized, resection can be performed via an incision through the 11th intercostal space on the affected side. If localization is unclear, exploration of both adrenal glands via abdominal or dorsal incisions is necessary. Resection of adrenal cortical adenomas is relatively straightforward, but adrenal cortical carcinomas often cannot be completely cured. Since the normal adrenal tissue outside the tumor is atrophic, corticosteroid supplementation is required both pre- and postoperatively. Postoperatively, ACTH 20r/d can be administered intramuscularly for 2 weeks to promote the recovery of atrophic cortical function. Hormone maintenance therapy should continue for more than 3 months postoperatively, followed by gradual tapering until discontinuation.
3. Bilateral Adrenalectomy Suitable for cases of bilateral adrenal cortical hyperplasia. The methods include: ① Total bilateral adrenalectomy: The advantage is rapid disease control and avoidance of recurrence; the disadvantage is lifelong corticosteroid supplementation postoperatively and an increased risk of Nelson's syndrome (pituitary tumor + hyperpigmentation). ② Total unilateral adrenalectomy with contralateral subtotal adrenalectomy: Since the right adrenal gland is closely adjacent to the inferior vena cava, reoperation for recurrent hyperplasia of residual adrenal tissue is extremely difficult. Therefore, total right adrenalectomy is generally performed. The residual left adrenal tissue should account for approximately 5% of the total adrenal weight. Excessive residual tissue increases the recurrence rate, while insufficient residual tissue or injury to the blood supply of the residual adrenal tissue may lead to adrenal cortical insufficiency or Nelson's syndrome. Thus, care must be taken during surgery to avoid injury to the blood supply. Since the adrenal blood supply is comb-like and distributed toward its edges, the residual tissue should be a small piece at the edge. Some authors advocate total unilateral adrenalectomy combined with pituitary radiation therapy, but this is often ineffective or associated with recurrence.
During adrenal surgery, the following points should be noted: ① Choice of incision: The 11th intercostal space incision can be used, but intraoperative position changes are required. Some patients with adrenal cortical adenoma diseases are misdiagnosed as adrenal cortical hyperplasia, leading to difficulties. For obese patients, bilateral adrenal exploration via the abdominal approach is challenging. A more suitable method is to place the patient in a prone position under general anesthesia and explore via a dorsal chevron incision (Nagamatsu incision, Figure 1) or through the 11th rib incision. Generally, the right side is explored first. If right adrenal hyperplasia (bilateral adrenal hyperplasia) or atrophy (left adrenal cortical adenoma) is found, the left adrenal gland should also be explored. If a right adrenal cortical adenoma is found, it can be excised without further exploration of the left side. Large adrenal carcinomas may require a thoracoabdominal combined incision (Figure 2). ② Corticosteroid supplementation: Patients with cortisol-secreting syndromes have high levels of cortisol secretion. A sudden drop in cortisol levels postoperatively can easily lead to an acute adrenal crisis due to adrenal insufficiency. Clinical manifestations include shock, tachycardia, tachypnea, cyanosis, nausea, vomiting, abdominal pain, diarrhea, high fever, unconsciousness, and even death. Therefore, corticosteroids should be supplemented preoperatively, intraoperatively, and postoperatively for prevention. Once a crisis occurs, rapid intravenous corticosteroid supplementation should be administered, along with correction of typical edema, electrolyte imbalances, and symptomatic treatment. Emotional fluctuations, infections, and certain surgical complications can trigger a crisis and sometimes confuse the diagnosis (e.g., pneumothorax, hemorrhage, etc.), so precautions should be taken to avoid such occurrences.
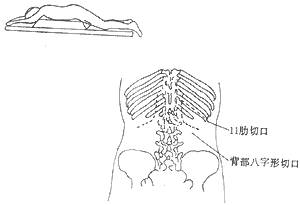
Figure 1 Exploration of the adrenal gland via the 11th rib and chevron incision on the back
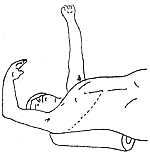
Figure 2 Exploration of a large adrenal tumor via a thoracoabdominal incision
Although the supplemented amount of corticosteroids exceeds the normal physiological secretion level, some cases may still experience a crisis due to the patient's preoperatively high cortisol secretion. Since postoperative crises mostly occur within 2 days after surgery, our hospital administers an additional intravenous dose of 100–200 mg/d hydrocortisone on the day of surgery and the following 2 days, significantly reducing the incidence of crises. If a crisis is suspected or surgical complications arise, the corticosteroid dosage should be increased. The long-term maintenance dose of corticosteroids is 25–37.5 mg/d cortisone acetate (the normal physiological requirement). Patients with adenomas generally need to maintain this dose for 3–6 months before discontinuation, while those who undergo bilateral adrenalectomy require lifelong medication. If the patient has other illnesses, infections, or undergoes procedures like tooth extraction, the hormone dosage should be increased. In cases of diarrhea or inability to eat, the medication should be switched to intramuscular administration. Patients should carry a medical certificate at all times for reference by doctors. Patients with adrenal adenomas or partial adrenalectomy can gradually discontinue medication once their condition stabilizes. To assess endogenous cortisol secretion levels before discontinuation, cortisone acetate can be stopped and replaced with dexamethasone (0.75 mg dexamethasone is equivalent to 25 mg cortisone acetate) for 1–2 weeks, followed by measurement of 24-hour urinary 17-hydroxy and 17-ketosteroid excretion. Since dexamethasone does not interfere with the measurement of urinary 17-hydroxy and 17-ketosteroids, the results reflect endogenous cortisol secretion levels. If the levels are close to normal, the dosage can be gradually reduced and discontinued. If the levels are very low, cortisone acetate should be resumed for maintenance. Some authors report that cutting the removed adrenal gland into small pieces and implanting them into the sartorius muscle or mesentery to treat postoperative adrenal insufficiency has shown some efficacy. Radioisotope-labeled cholesterol scans confirm radioactive concentration in the transplant area, and urinary 17-hydroxycorticosteroid excretion also increases, allowing some patients to reduce or discontinue corticosteroid maintenance. If hyperfunction occurs, a minor local surgery can be performed to remove the tissue. Due to the small size of adrenal stirred pulses, vascularized autologous adrenal transplantation is challenging. ③Management of Nelson’s syndrome: After total adrenalectomy, preexisting pituitary adenomas or microadenomas may continue to grow, compressing the optic nerve and causing visual impairment. Pituitary-secreted melanocyte-stimulating hormone leads to generalized skin and mucous membrane pigmentation, sometimes turning bronze. Pituitary adenomectomy can restore vision, and localized pituitary radiotherapy can inhibit tumor growth. Traditional Chinese medicine and herbs also have some efficacy in alleviating pigmentation.
II. Non-surgical therapy
1. Pituitary radiotherapy: 20% of cases achieve lasting efficacy. However, most cases respond poorly and relapse easily, so it is generally not the first choice. Before pituitary radiotherapy, it must be confirmed that there is no adrenal tumor.
Table 2 Routine corticosteroid supplementation protocol
| Hydrocortisone (intravenous) | Cortisone acetate (intramuscular) | Cortisone acetate (oral) | |
| 2 days preoperatively 5 | 0mg, every 8 hours | ||
| Postoperative Day 1 (Surgery Day) | 100mg (intraoperative) | 50mg, every 8 hours | |
| Postoperative Day 2 | 50mg, every 8 hours | ||
| Postoperative Day 3 | 50mg, every 8 hours | ||
| Postoperative Days 4-5 | 50mg, every 8 hours | ||
| Postoperative Day 6 | 25mg, every 8 hours | ||
| Postoperative Day 7 | 25mg, three times daily | ||
| Postoperative Days 8-9 | 25mg, twice daily | ||
| From Postoperative Day 10 | 12.5mg, three times daily |
2. Drug therapy has significant side effects and uncertain efficacy, mainly applicable to cases of unresectable adrenal cortical adenocarcinoma. ① Dichlorodiphenyldichloroethane (O,P´DDD): Causes necrosis of adrenal cortical zona reticularis and zona fasciculata cells. Suitable for metastatic and incurable functional or non-functional cortical carcinoma. However, it has severe gastrointestinal and neurological side effects and may lead to acute adrenal cortical insufficiency. The treatment dose ranges from 4 to 12g/d, starting with a small dose and gradually increasing to a maintenance dose, adjusted based on the patient's tolerance and cortical function. ② Metyrapone (Su4885): An 11β-hydroxylase inhibitor. It inhibits the conversion of 11-deoxycortisol to cortisol and 11-deoxycorticosterone to corticosterone, thereby reducing cortisol synthesis. Side effects are minor, mainly gastrointestinal reactions. However, its effects are temporary and only alleviate symptoms. Once cortisol secretion decreases, it stimulates ACTH secretion, overcoming its blocking effect. ③ Aminoglutethimide: Inhibits cholesterol synthesis into pregnenolone. For mild adrenal cortical hyperplasia, 1–1.5g/d is administered, while severe cases require 1.5–2g/d to control symptoms. Close monitoring of corticosteroid levels is necessary, and small doses of glucocorticoids and mineralocorticoids should be supplemented if needed to prevent adrenal cortical insufficiency. ④ Cyproheptadine: A serotonin competitor. Since serotonin stimulates the hypothalamic-pituitary axis to release ACTH, cyproheptadine can inhibit pituitary ACTH secretion. It is suitable for treating bilateral adrenal hyperplasia. The dose is gradually increased from 8mg/d to 24mg/d. After total or subtotal adrenalectomy, while supplementing corticosteroids, taking cyproheptadine can reduce the chance of pituitary tumor occurrence. Other drugs such as bromocriptine and trilostane have also been reported to have certain efficacy.
Patients with cortisol syndrome, if left untreated, rarely recover spontaneously, and the general course of the disease does not exceed 5 years. The causes of death include infections, {|###|}heart and blood vessel diseases, uremia, gastrointestinal bleeding, diabetic unconsciousness, cancer metastasis, etc.
The 5-year survival rate for bilateral adrenal hyperplasia treated with bilateral adrenal surgery or pituitary tumor resection can reach approximately 85-95%. However, outcomes are poorer with only pituitary radiotherapy or unilateral adrenal surgery. The best therapeutic results are achieved with adenoma resection. The prognosis for adrenal cancer is very poor, with a 5-year survival rate of only 10% after extensive surgery. For cases of cancer recurrence or ectopic ACTH syndrome treated with O,P´DDD, the 5-year survival rate is zero.


