| disease | Congenital Aortic Arch Anomalies |
Congenital aortic arch anomalies refer to developmental abnormalities of the aortic arch and its branches, leading to compression of the trachea and/or esophagus. Hommel described the double aortic arch in 1737. In 1939, Wolman detailed the clinical manifestations of tracheal and esophageal compression caused by a double aortic arch. Gross performed the first surgical treatment for a double aortic arch in 1945, which advanced the discovery and understanding of various types of aortic arch anomalies. Diagnostic techniques and treatment methods have since developed and improved, yielding favorable outcomes. Among congenital cardiovascular anomalies, aortic arch and branch anomalies account for only 1–2%.
bubble_chart Pathogenesis
During the fourth week of embryonic development, the anterior ends of the bilateral dorsal aortae curve around the pharyngeal gut and form the first pair of aortic arches and the left and right primitive aortae on the ventral side of the foregut. The latter fuse to form the aortic sac. As the branchial arches grow, six pairs of branchial aortic arches successively emerge from the aortic sac and connect with the dorsal aortae. When the third pair of branchial aortic arches fully develops, the first and second pairs of branchial aortic arches disappear. The third pair of branchial aortic arches forms the common carotid artery and part of the internal carotid artery. The fourth pair of branchial aortic arches forms the aortic arch on the left side and the innominate artery and the right subclavian artery trunk on the right side. The fifth pair of branchial aortic arches either does not consistently exist or disappears quickly. The sixth pair of branchial aortic arches forms the pulmonary artery, with its distal segment on the right side disconnecting from the dorsal aorta. On the left side, it persists during the fetal period and is called the ductus arteriosus, which closes after birth to become the ligamentum arteriosum (Figure 4).
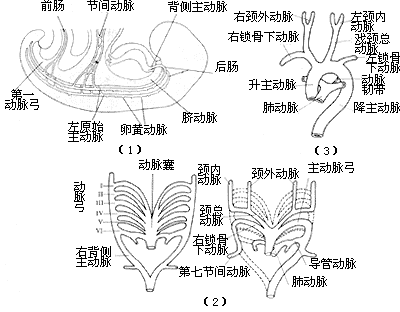
Figure 4 Normal Embryology of the Aortic Arch
If abnormalities occur during the development of the branchial aortic arches or the dorsal aortae, various malformations of the aortic arch and its branches may result after birth. Most cases involve only malformations of the aortic arch or its branches, while a minority may coexist with other cardiac anomalies such as tetralogy of Fallot or transposition of the great arteries. Based on the developmental status of the fourth branchial aortic arch and the branches of the aortic arch, the position of the descending aorta, and the course of the ductus arteriosus or ligamentum arteriosum, abnormalities of the aortic arch and its branches can be classified into the following types.
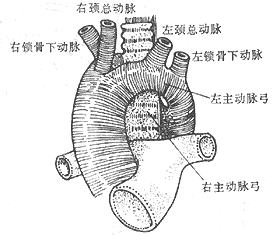
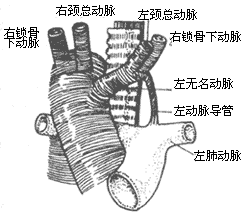
Double Aortic Arch: If both fourth branchial aortic arches persist and develop, a double aortic arch is formed. The ascending aorta is normal and divides into left and right aortic arches outside the pericardial membrane. The left aortic arch runs from right to left in front of the trachea, crosses over the left main bronchus, and merges with the right aortic arch on the left side of the spine to form the descending aorta. The right aortic arch crosses over the right main bronchus, passes behind the esophagus in front of the spine, and descends leftward across the midline to merge with the left aortic arch to form the descending aorta. Each aortic arch gives off two branches: the left aortic arch gives rise to the left common carotid artery and the left subclavian artery, while the right aortic arch gives rise to the right common carotid artery and the right subclavian artery. The ductus arteriosus or ligamentum arteriosum is located between the lower edge of the left aortic arch near the origin of the left subclavian artery and the left pulmonary artery. In most cases, the diameters of the two aortic arches are unequal, with the right side usually being larger (Figure 5).
(1) Frontal View
(2) Lateral View
Figure 5 Double Aortic Arch
In rare cases, the descending aorta is located on the right side. The left aortic arch crosses over the left main bronchus, then passes posteriorly and rightward behind the esophagus, merging with the right aortic arch on the right side of the spine to form the descending aorta. Regardless of whether the descending aorta is on the left or right, the vascular ring formed by the double aortic arch surrounds the trachea and esophagus. If the space between the two aortic arches is narrow, clinical symptoms of compression may occur.
Sometimes the distal segment of the left aortic arch may become occluded, resembling a fibrous band. The occlusion may occur between the left common carotid artery and the left subclavian artery, between the left subclavian artery and the ductus arteriosus or ligamentum arteriosum, or between the ductus arteriosus (ligamentum arteriosum) and the merging point of the left and right aortic arches (Figure 6).
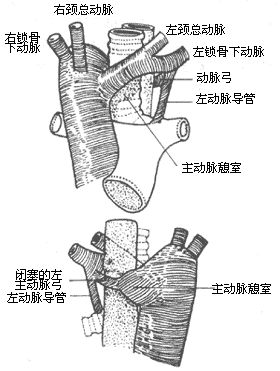
Figure 6 Double Aortic Arch with Left Arch Occlusion
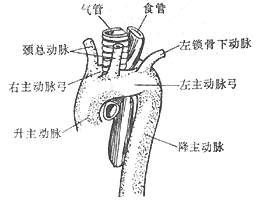
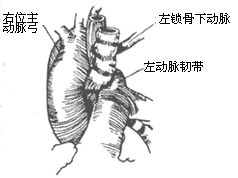
Right-sided aortic arch: The left fourth aortic arch degenerates and disappears, while the right side develops to form the main aortic arch, with the descending aorta located on the right side of the spine. The branching sequence from the main aortic arch is a mirror image of the normal arrangement, i.e., the first branch is the left innominate artery, which then gives off the left common carotid artery and the left subclavian artery; the second branch is the right common carotid artery; and the third branch is the right subclavian artery. Sometimes, the main aortic arch gives off four branches, with the left innominate artery absent, and the arterial duct or ligamentum arteriosum located between the left innominate artery or left subclavian artery and the left pulmonary artery. The absence of vessels behind the esophagus does not constitute a vascular ring. A right-sided aortic arch generally does not compress the trachea or esophagus, but in rare cases, the arterial duct or ligamentum arteriosum may pass behind the esophagus from the left pulmonary artery to connect to the distal segment of the right-sided aortic arch, or the left subclavian artery may originate from the proximal segment of the descending aorta and pass behind the esophagus to reach the left upper limb. The arterial duct or ligamentum arteriosum may also be located between the left pulmonary artery and the left subclavian artery on the left side of the trachea, or between the left pulmonary artery and the left subclavian artery originating from the descending aorta. In these cases, if the arterial duct or ligamentum arteriosum is short, it may form part of a vascular ring, causing symptoms of tracheal or esophageal compression (Figure 7).
(1) Right-sided aortic arch stirred pulse:
(2) Right-sided aortic arch stirred pulse with left subclavian stirred pulse causing compression
Figure 7: Right-sided aortic arch stirred pulse
Left-sided aortic arch stirred pulse: The left-sided aortic arch stirred pulse rarely forms a vascular ring. In some cases, the left subclavian stirred pulse has an anomalous origin from the distal aortic arch stirred pulse and passes behind the esophagus to enter the right upper limb. Sometimes, the stirred pulse ductus or stirred pulse ligament connects the anomalous right subclavian stirred pulse to the right pulmonary stirred pulse. Additionally, a left-sided aortic arch stirred pulse with a right-sided descending aorta stirred pulse, where the left aortic arch stirred pulse loops behind the esophagus to connect to the descending aorta stirred pulse on the right side, may involve the stirred pulse ductus or stirred pulse ligament in the formation of a vascular ring. Alternatively, an anomalous left subclavian stirred pulse may arise from the proximal descending aorta stirred pulse, passing behind the esophagus to enter the left upper limb. These conditions can all form vascular rings, leading to compression symptoms (Figure 8).
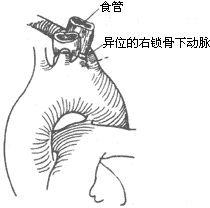
Figure 8: Left-sided aortic arch stirred pulse with anomalous right subclavian stirred pulse compressing the esophagus
Anomalous innominate stirred pulse: The aortic arch stirred pulse and its branches develop normally, but the innominate stirred pulse originates from the aortic arch stirred pulse more to the left side, looping upward and to the right around the anterior wall of the trachea into the right thoracic apex. If the innominate stirred pulse is long and loose, it may not cause symptoms. However, if the vessel is large, short, and tight, it can severely compress the trachea.
Anomalous aberrant left pulmonary stirred pulse: Glaevecke and Doehle first reported in 1897 an anomalous aberrant left pulmonary stirred pulse originating from the posterior aspect of the right pulmonary stirred pulse. It then crosses over the right main bronchus and turns leftward, entering the left hilum between the trachea and esophagus. This forms a sling around the right main bronchus and the lower segment of the trachea, causing compression symptoms and potentially associated with hypoplasia of the lower trachea and right main bronchus. Half of the patients exhibit symptoms at birth, and about two-thirds of cases show symptoms within the first month of life. The most common symptom is respiratory obstruction, with wheezing during expiration, often accompanied by recurrent episodes of acute respiratory obstruction and infections. However, dysphagia is not typically present.
Chest X-ray may reveal differences in lung translucency between the two sides, with hyperinflation of the right lung. A plain chest film may show a shadow of the anomalous left pulmonary stirred pulse in the carinal region between the trachea and esophagus. The branches of the left pulmonary stirred pulse in the left hilum may appear lower in position. Bronchoscopy or bronchography may reveal associated tracheobronchial anomalies, but these procedures carry the risk of worsening respiratory obstruction. Chest fluoroscopy may detect differences in tracheal width between the two sides, and sometimes pulmonary stirred pulse angiography can confirm the diagnosis.
bubble_chart Clinical Manifestations
The main aortic arch and its branch anomalies themselves do not affect circulatory physiology or hemodynamics. However, if a vascular ring or a vascular ring combined with fibrous bands or ectopic aortic arch branches compresses the trachea or esophagus, clinically varying degrees of respiratory compression and/or dysphagia may occur, with severe cases potentially leading to death.
In cases where tracheal or esophageal compression is severe, symptoms such as inspiratory stridor with expiratory wheezing, tachypnea, coarse breath sounds, persistent cough, and hoarse crying may appear immediately after birth. Sometimes, dyspnea, cyanosis, brief respiratory pauses, or loss of consciousness may occur. Symptoms of dyspnea worsen during feeding or lying supine and may alleviate when lying on the side or extending the head and neck. Severe respiratory compression may manifest as marked retraction of the supraclavicular fossa and subcostal margins during inspiration. Recurrent respiratory infections are common, with worsening airway obstruction during episodes. In cases of esophageal compression, refusal to eat and dysphagia often occur, with frequent choking during meals accompanied by vomiting, aggravated dyspnea, and poor nutritional development.
In most cases, the aforementioned tracheal and esophageal compression symptoms begin to appear within the first 6 months after birth. Severe compression may present symptoms within days of birth. Without treatment, such cases often prove fatal before the age of one. In milder cases where symptoms appear after 6 months without progressive worsening, symptoms may gradually resolve during growth but worsen again during respiratory infections. Dysphagia caused by compression of the esophagus by an ectopically originating subclavian artery may only manifest in middle age (around 40 years) when the ectopic artery develops sclerotic changes and enlarges. Occasionally, an ectopic subclavian artery may develop aneurysmal dilation.
Chest X-ray: In cases without other congenital heart anomalies, the cardiac silhouette on plain chest radiographs may appear normal. In cases of double aortic arch, bilateral spherical bulges of the aortic arch can be observed, with the right side being more prominent. Barium esophagography may reveal indentations on both sides of the upper esophagus at the level of the third and fourth thoracic vertebrae. The indentation caused by the right aortic arch is larger and located higher, while that caused by the left aortic arch is smaller and lower. Tomography may show signs of tracheal compression. In cases of right-sided aortic arch, the chest X-ray only shows a spherical bulge of the aortic arch on the right side, with absence on the left. Barium esophagography in such cases reveals the esophagus being pushed to the left with visible indentations. In cases of aberrant subclavian artery, esophagography may show oblique or spiral indentations on the posterior esophageal wall due to vascular compression. For infant cases, iodized oil or water-soluble contrast agents are preferred for esophagography, as barium contrast agents, if aspirated into the tracheobronchial tree, may worsen dyspnea or lead to aspiration pneumonia.
Bronchoscopy: Bronchoscopy can identify the site of tracheal compression and allow observation of vascular pulsations at the compressed area. However, it must be performed with extreme caution, as trauma and edema of the respiratory mucosa may exacerbate airway obstruction.
Aortography: Aortography is the most reliable diagnostic method for confirming aortic arch and branch anomalies. A catheter is inserted into the ascending aorta, and contrast medium is injected to visualize the aorta and its branches. Biplane cineangiography can reveal the origin, course, caliber, and other abnormalities of the aortic arch and its branches, thereby establishing a definitive diagnosis.
bubble_chart Treatment Measures
In cases where the main aortic arch and its branch anomalies produce significant symptoms of respiratory tract and esophageal compression, surgical treatment should be performed. Depending on the specific condition of the lesion, the vessels or fibrous cord-like tissues, including the ligamentum arteriosum, that cause compression of the trachea and esophagus should be cut or freed to fully release the trachea and esophagus and eliminate the symptoms.
Preoperative use of antibiotics is required to control respiratory infections, clear respiratory secretions, and improve overall condition through fluid infusion and nasogastric feeding to enhance nutrition.
The anesthesia method of choice is endotracheal intubation anesthesia, and maintaining airway patency should be ensured throughout the entire surgical procedure.
The most commonly used surgical incision is a left posterolateral thoracotomy, entering the chest through the fourth intercostal space. The method for handling the vascular ring depends on the specific condition of the lesion.
Double aortic arch: If the distal segment of the left aortic arch is thinner and the ligamentum arteriosum is located on the left side, after entering the chest, the mediastinal pleura is incised behind the vagus nerve or between the vagus nerve and the phrenic nerve, and the recurrent laryngeal nerve is freed, taking care to avoid injury to the recurrent laryngeal nerve and the thoracic duct. The ductus arteriosus or ligamentum arteriosum is dissected and freed, then ligated and cut or sutured after cutting. The distal segment of the left aortic arch is separated, and non-traumatic vascular clamps are placed between the origin of the left subclavian artery and the descending aorta or between the left common carotid artery and the left subclavian artery. The distal segment of the left aortic arch is then cut, and the proximal and distal ends are sutured separately. After fully freeing the proximal end of the left aortic arch, the cut end is sutured and fixed to the anterior chest wall fascia. The fibrous tissue around the trachea and esophagus is then stripped to fully release the trachea and esophagus, and the mediastinal pleura does not need to be sutured (Figure 1).
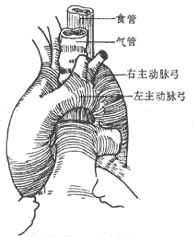
(1) Shows a double aortic arch
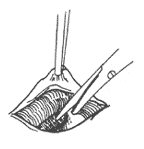
(2) Ligation and cutting of the ligamentum arteriosum

(3) Clamping the left aortic arch
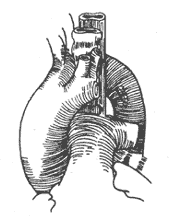
(4) The left aortic arch has been cut
Figure 1 Double aortic arch, cutting the ductus and left arch
In cases where the left aortic arch has a larger diameter, after ligating and cutting the ligamentum arteriosum, the distal segment of the left aortic arch and the descending aorta are dissected and freed, a ligature is placed around them and pulled to the left to expose the distal segment of the right aortic arch. Non-traumatic vascular clamps are then placed, and the left aortic arch is cut. The two cut ends are carefully sutured, and the trachea and esophagus are fully freed from the mediastinal fibrous tissue and the left aortic arch. The left aortic arch and descending aorta are then sutured and fixed to the anterior chest wall fascia or the periosteum of the rib.
Right-sided aortic arch, left-sided ligamentum arteriosum, and retroesophageal aberrant left subclavian artery: The vascular ring formed in this situation can be addressed by entering the left chest to ligate and cut the ligamentum arteriosum, cutting and ligating the right subclavian artery on the left side of the esophagus, and after anatomically separating the descending aorta and esophagus, suturing and fixing them to the chest wall fascia. To prevent postoperative subclavian steal syndrome, the distal cut end of the right subclavian artery can be anastomosed end-to-side to the left common carotid artery or the aortic arch, and the vertebral artery can also be ligated simultaneously.
Left main artery arch, esophageal posterior ectopic right subclavian artery: This condition usually only compresses the esophagus, causing dysphagia symptoms. Enter the chest through a left posterolateral incision at the 4th intercostal space, free the right subclavian artery near the main artery arch, place a non-traumatic vascular clamp to cut and suture the right subclavian artery, then free the distal segment of the right subclavian artery and push it to the right side of the esophagus, while freeing and cutting the fibrous tissue around the esophagus. To prevent subclavian artery steal syndrome, the vertebral artery can be ligated simultaneously. For older cases, a right posterolateral thoracotomy incision can be used to cut the right subclavian artery, suture the proximal incision, and perform an end-to-side anastomosis between the distal segment of the right subclavian artery and the right common carotid artery or the main artery arch (Figure 2).
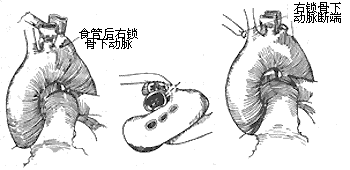
Figure 2 Left aortic arch with aberrant right subclavian artery
Innominate artery anomaly: Through a right or left fourth intercostal anterior thoracotomy, the innominate artery is mobilized, and the innominate artery along with the aortic arch is sutured and fixed to the anterior chest wall to relieve tracheal compression.
The treatment for aberrant left pulmonary artery involves entering the chest through a left posterolateral thoracotomy at the third or fourth intercostal space. After mobilizing the trachea, esophagus, hilum, and left pulmonary artery, the arterial ligament is ligated and divided. The left pulmonary artery is then dissected posterior to the left bronchus, transected near its origin from the right pulmonary artery, and the right pulmonary artery incision is sutured. The left pulmonary artery is pulled out from behind the trachea and anastomosed end-to-end with the main pulmonary artery anterior and to the left of the trachea (Figure 3). Early postoperative mortality is high, primarily due to grade III tracheobronchial stenosis and complications such as kinking or thrombosis of the left pulmonary artery caused by improper anastomosis.
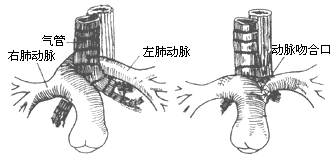
Figure 3 Aberrant left pulmonary artery
Postoperative management: In cases of aortic arch and its branch anomalies, the tracheal cartilage rings, which are underdeveloped and weak due to compression, are prone to collapse during inspiration. Therefore, continuous endotracheal intubation with positive pressure and high-humidity oxygen inhalation is required for several days postoperatively. Secretions should be suctioned to maintain airway patency. Intravenous administration of a small dose of dexamethasone can reduce tracheal mucosal edema after extubation. In some cases, tracheal and esophageal compression symptoms may take weeks or months to completely resolve postoperatively.
The current surgical mortality rate has been reduced to 5-10%. Cases without other congenital heart and vascular malformations have good long-term outcomes.


