| disease | Pain Wind |
| alias | Gout |
Pain wind (gout) is a disease caused by hereditary or acquired sexually transmitted diseases that lead to purine metabolism disorders and persistently elevated serum uric acid levels. Currently, cases of pain wind have been reported worldwide. Since the first domestic report in 1948, the incidence of this disease has gradually increased with the improvement of living standards and changes in dietary structure, particularly in high-altitude pastoral areas, Qinghai, and Tibet.
bubble_chart Epidemiology
The incidence of pain wind varies significantly around the world. Among adult Maori males in New Zealand, the incidence rate is as high as 8%, while among the white population in New Zealand, it is only 0.5%. Factors influencing its onset, besides race, include genetics, protein content in the diet, social life, cultural conditions, and mental stress.
The incidence of primary pain wind shows a very significant gender difference. The author's statistics from 879 reported cases in the country show that 807 cases were male, accounting for 91.8%, and 72 cases were female, accounting for 8.2%. However, the incidence rate in women increases after menopause, accounting for about 5-10% of all cases.
The incidence of pain wind has a significant age characteristic. Although it can be seen in all age groups, primary pain wind is most common in middle-aged people, with the peak incidence between 40 and 50 years old. The average age of onset is 44 years. The incidence rate in people over 60 years old accounts for 11.6% of all cases, with a relative increase in women to 29%. In children and elderly pain wind patients, the incidence of secondary pain wind is higher, so attention should be paid to distinguishing between primary and secondary pain wind in children and elderly patients.
The incidence of primary pain wind is closely related to the protein content in the diet. During the First and Second World Wars, due to the decline in diet quality, the incidence of pain wind in Europe significantly decreased. When the protein content in the diet became abundant again after the war, the incidence rate returned to pre-war levels. After Japan's economic boom in the 1960s, the protein content in the national diet significantly increased, making pain wind a more common disease among Japanese people. Clinical reports of pain wind in the country have also increased year by year, especially since the 1980s, with a significant increase.The prerequisite for the onset of gout is hyperuricemia. At a blood pH of 7.4, uric acid in the blood exists in the form of sodium urate ions, hence hyperuricemia is also known as hyperuricosemia. All clinical manifestations of gout are caused by the precipitation and deposition of its sodium salt from supersaturated extracellular fluid into tissues. In addition to the crystallization of urate, kidney lesions in gout can also be caused by the crystallization of uric acid itself in a few cases, such as acute uric acid nephropathy. Many uric acid kidney stones are also caused by uric acid crystallization.
I. Causes of Hyperuricemia
The serum sodium urate level in normal individuals fluctuates within a narrow range. The average value for normal males in China is 339 μmol/L (5.7 mg/dl), and for females, it is 256 μmol/L (4.3 mg/dl). The upper limit of normal uric acid for males is 417 μmol/L (7.0 mg/dl), and for females, it is 357 μmol/L (6.0 mg/dl). The level of uric acid in the blood is influenced by factors such as race, diet, habits, age, weight, and body surface area. Generally, uric acid levels increase with age, especially more pronounced in women after menopause. Therefore, clinically, hyperuricemia is defined as exceeding the above normal standards or being more than 2 standard deviations (SD) above the average for the same gender.
(1) Balance of Uric Acid MetabolismThe concentration of uric acid in serum depends on the balance between the rate of uric acid production and excretion. Uric acid is the end product of purine metabolism. The accumulation of uric acid in the body is seen in the following five situations: ① Increased exogenous absorption, i.e., increased intake of purine-rich foods; ② Increased endogenous biosynthesis, including enzyme defects, such as accelerated nucleic acid decomposition and increased uric acid from purine base oxidation products; ③ Reduced excretion, i.e., reduced excretion through the kidneys in urine and reduced decomposition by intestinal cells after secretion by the gallbladder and gastrointestinal tract; ④ Reduced metabolism in the body, i.e., reduced endogenous destruction of uric acid; ⑤ A combination of the above factors or different factors.
Due to the lack of uricase in human tissues, uric acid cannot be decomposed. The amount of uric acid degraded to allantoin and carbon dioxide by peroxidase in human leukocytes is limited, so endogenous uric acid decomposition is also secondary. Therefore, increased endogenous purine production and reduced uric acid excretion, or a combination of both, are very important in the pathogenesis of both primary and secondary hyperuricemia.
(2) Causes of Uric Acid Accumulation
The main cause of uric acid accumulation is increased endogenous purine production, with reduced uric acid excretion accounting for a minority. When American male citizens are on a purine-restricted diet, the normal range of uric acid excretion is 1.48~3.54 mmol/d (250~600 mg/d). Most primary gout patients have a 24-hour urinary uric acid excretion within the normal range, with 20~25% of patients having increased excretion. Studies on the metabolic turnover rate of the uric acid pool and glycine tracing have shown that patients with increased urinary uric acid excretion have abnormally increased purine synthesis, while 70~78% of patients with normal urinary uric acid excretion have normal excretion, and only 2% have excessively low excretion. Gutman et al.'s analysis of 300 primary gout cases showed that one-third had both increased uric acid production and reduced excretion.
II. The Role of Enzyme Defects in PathogenesisEnzyme defects include an increase in the quantity or activity of certain enzymes and the complete or partial lack of others. Both can lead to accelerated purine synthesis and increased uric acid production. Enzyme defects play a very important role in the pathogenesis of gout, but most are difficult to confirm, with only a few patients being able to be identified with enzyme defects.
(1) Regarding Heredity
Primary hyperuricemia-induced gout is mostly hereditary. However, clinically, only 10-20% of gout patients have a family history of the disease. This is related to the polygenic, dominant, and recessive inheritance patterns. Type I glycogen storage disease is autosomal recessive; hypoxanthine phosphoribosyltransferase (HGPRT) deficiency, phosphoribosyl pyrophosphate (PPRP) synthetase structural abnormalities, and epidemic hyperuricemia are X-linked recessive; primary gout caused by incomplete HGPRT deficiency is transmitted by female carriers and manifests in males; postmenopausal female gout is associated with HGPRT deficiency.
(II) Research Progress on Enzyme Deficiencies
With the in-depth development of basic medicine and clinical research, current understanding of the molecular defects leading to excessive purine biosynthesis has improved. Changes in enzyme quantity, structural abnormalities, and enhanced activity can all accelerate purine synthesis, thereby increasing uric acid production.
1. Increase in Enzyme Quantity and Activity In primary gout, there are four types caused by an increase in enzyme quantity and activity.
(1) Excessive Glutathione Reductase: An excess of this enzyme catalyzes the conversion of reduced nicotinamide adenine dinucleotide phosphate (NADPH) and oxidized glutathione (GSSG) into nicotinamide adenine dinucleotide phosphate (NADP) and reduced glutathione (GSH). NADP is a coenzyme of the pentose phosphate pathway, and its excess promotes this pathway, thereby increasing the synthesis of 5-phosphoribose, which is the substrate for phosphoribosyl pyrophosphate (PRPP). PRPP is an important precursor for purine biosynthesis, so an increase in this precursor also leads to increased uric acid production.
(2) Increase in Quantity and Activity of Glutamine Phosphoribosyl Pyrophosphate Amidotransferase (GPR-PPAT): In the process of purine synthesis, GPRPPAT catalyzes the formation of 1-amino-5-phosphoribose (PRA), a key reaction. An increase in GPRPPAT quantity or activity promotes PRA production, leading to increased synthesis of inosine monophosphate and, consequently, increased uric acid production.
(3) Increased Activity of Phosphoribosyl Pyrophosphate (PRPP) Synthetase: Increased PRPP synthetase activity promotes the synthesis of nucleic acids and purine bases, leading to increased uric acid production.
(4) Increased Activity of Xanthine Oxidase (XO): Increased XO activity accelerates the oxidation of hypoxanthine to xanthine, further accelerating the production of uric acid from xanthine. The increase in XO activity is due to secondary hepatic enzyme induction rather than a congenital defect.
2. Enzyme Deficiency or Reduced Activity Complete deficiency, partial deficiency, or reduced activity of enzymes can also lead to increased purine base synthesis. The following four types have been clearly identified.
(1) Glucose-6-Phosphatase Deficiency: This enzyme deficiency causes type I glycogen storage disease. 6-phosphate glucose cannot be converted into glucose, and metabolism shifts to phosphogluconate, which is partially converted into 5-phosphoribose. This serves as a raw material for nucleic acid synthesis, leading to hyperactive nucleic acid synthesis and increased production of purines and uric acid.
(2) Glutaminase Deficiency: Deficiency of this enzyme reduces the breakdown of glutamine, leading to its accumulation and an increase in substrates for purine base synthesis, resulting in increased uric acid production.
(3) Reduced Activity of Glutamate Dehydrogenase: Reduced activity of this enzyme decreases the production of α-ketoglutarate from glutamyl dehydrogenation, shifting towards increased glutamine, which enhances purine and uric acid synthesis.
(4) Hypoxanthine-Guanine Phosphoribosyltransferase (HGPRT) Deficiency: HGPRT deficiency impairs purine recycling, leading to purine base accumulation and increased uric acid production.
III. Causes of Urate Deposition in Tissues
The majority of urates in the blood are sodium urate, and urates in a dissolved state are non-toxic. Urate is present in all tissues except the central nervous system. Excessive deposition of urate in tissues forms gouty tophi. Physical effects cause mechanical injury to tissues, leading to tissue rupture and destruction, which triggers an inflammatory response. When urate deposits in the synovial membrane or synovial cavity, it appears as microcrystals, which are the basis for acute inflammatory reactions.
(I) Issues Regarding Urate Deposition in Tissues
Sodium urate in the blood, at a body temperature of 37℃, is measured in μmol/L (6.4mg/dl), with a small portion bound to protein (24μmol/L, 0.4mg/dl). Exceeding this limit results in a supersaturated state. Sodium urate in a saturated state, when bound to plasma-specific α1 and α2 globulins, still maintains a certain degree of stability. If the concentration of sodium urate is excessively high or the levels of plasma-specific α1 and α2 globulins are reduced, the bound sodium urate is prone to crystallize and precipitate in tissues. Some individuals have serum sodium urate levels as high as 535μmol/L (9.0mg/dl), persisting for many years or even a lifetime, yet they do not exhibit symptoms of gout, which may be related to this. Others believe that the precipitation of urate is related to a decrease in the amount of serum albumin binding. Some scholars suggest that urate bound to protein accounts for only 4-5% in normal plasma, and whether this is of significant importance requires further research.
(II) On the issue of uric acid deposition in urine
Uric acid exists in urine in the form of dilute alcohol sodium and potassium salts. Its solubility is related to the pH of the urine. As the pH of the urine decreases, it shifts towards free uric acid. The titration curve of urine indicates that at pH 5.0, 85% of the uric acid is undissociated, and only 15 mg of urate can be dissolved per 100 ml of urine. Most of the undissociated uric acid is deposited in the form of microcrystals. If a large amount is deposited in the distal collecting tubules, it can cause tubular obstruction, leading to acute renal failure. Deposition in the renal pelvis forms crystalluria; if there is also a significant amount of protein matrix present, uric acid kidney stones can form. Therefore, acidic urine is very unfavorable for the excretion of urates. If the urine pH is 7.0, the solubility of uric acid can be more than 10 times that at pH 5.0. Hence, the precipitation of uric acid in urine is critically related to the pH level of the urine. Additionally, factors such as the total daily urine volume, the total amount of urate excreted, and the content of other stabilizing substances in the urine also affect the deposition of uric acid.
IV. Urate Metabolism in the Kidney
The metabolism of urates in the kidney includes three processes: glomerular filtration, tubular reabsorption, and resecretion. Any disruption in these processes can lead to hyperuricemia.
(I) Normal Metabolic Process
About 92-95% of the urates in the blood can pass through the glomerular basement membrane, with only 5-8% bound to non-diffusible components circulating in the blood. About 90-98% of the urates filtered through the basement membrane are reabsorbed by the proximal tubule and the loop of Henle. The urates excreted in the urine, about 80-85%, come from the proximal and distal tubules after resecretion and are partially reabsorbed a second time.
(II) Metabolic Defects in Pain Wind Patients
Pain wind patients show reduced 24-hour uric acid excretion, indicating a defect in the tubular secretion of urates, possibly due to reduced blood flow to the tubules, decreased rate of urate entry into tubular cells, or a defect in the secretion transport system. Only when the hematuria urate content is 119-178 μmol/L (2-3 mg/dl) higher than the normal value can the same excretion rate be achieved.
(III) The Impact of Water and Electrolyte Metabolism Disorders on Urate Excretion
The transport of urates in the proximal tubule is closely related to water and sodium metabolism balance. Dehydration, sodium deficiency, use of diuretics, diabetes insipidus, etc., cause insufficient blood volume, leading to increased reabsorption of sodium and water in the proximal tubule, and consequently increased reabsorption of uric acid, resulting in elevated serum urate concentration. Sodium retention, pregnancy, and excessive secretion of antidiuretic hormone increase blood volume, reducing urate reabsorption, increasing clearance, and lowering serum urate concentration.
(IV) Competition of Organic Acids for Urate Transport
Certain organic acids can competitively inhibit the excretion of urates. For example, in lactic acidosis and ketoacidosis, increased excretion of lactic acid, β-hydroxybutyric acid, and acetoacetic acid competitively inhibits urate excretion, significantly increasing serum urate concentration. Some uricosuric drugs like probenecid, also organic acids, can inhibit both the secretion and reabsorption of urates in the renal tubules. Small doses can inhibit urate resecretion, causing urate retention; larger doses have a uricosuric effect. Aspirin similarly inhibits resecretion at small doses, causing urate retention; at large doses, it hinders renal tubular reabsorption of urates, thereby increasing urate excretion.
(V) The Impact of Renal Disease on Urate Excretion
Renal parenchyma sexually transmitted diseases such as kidney inflammation, heart blood vessel diseases, and hypertension leading to renal insufficiency can cause a decrease in the glomerular filtration rate, which in turn can lead to an increase in serum uric acid levels. This is followed by a compensatory increase in renal tubular secretion and gastrointestinal excretion. However, when the glomerular filtration rate decreases to <25ml/min, these compensatory functions lose their effectiveness. Therefore, in patients with uremia, serum uric acid levels are significantly elevated. In some diseases that primarily damage the renal tubules, such as polycystic kidney disease and lead poisoning, the increase in serum uric acid levels is mainly due to a decrease in renal tubular re-secretion.
V. The Mechanism of Acute Arthritis
The formation of sodium urate microcrystals in the joint cavity, leading to nonspecific joint inflammation, is a complex process that may result from the combined effects of multiple factors.
(I) Formation of Sodium Urate Microcrystals
When the concentration of sodium urate in the blood or synovial fluid reaches a saturated state, crystal precipitation occurs. Therefore, the onset of acute gouty arthritis is positively correlated with the degree of hyperuricemia. However, many patients with hyperuricemia never experience an acute arthritis attack in their lifetime. Some patients only experience pain wind after years of sustained hyperuricemia. In contrast, a few patients with acute pain wind have significantly lower uric acid concentrations than the saturation state. Additionally, some patients experience acute pain wind after uric acid-lowering treatment, known as urate migratory attacks. The mechanism may be related to the following factors.
1. Proteoglycan Theory: Roberts believes that cartilage and synovial fluid contain various proteoglycans. Each proteoglycan molecule not only occupies a large space but also carries a large number of negative charges. The anion gap of proteoglycans can significantly increase the solubility of sodium urate, thereby inhibiting the formation of crystals. If the molecular structure of proteoglycans is incomplete or digested by trypsin, the solubility of urate decreases, reducing the ability to inhibit microcrystal formation, which may lead to acute pain wind attacks.
2. Temperature-Related Theory: There is a certain gradient between the core body temperature and the temperature in the distal limbs and peripheral joint cavities. For example, the temperature of the toes and ear margins is significantly lower than the core body temperature. Some have measured the temperature inside the knee joint cavity to be about 32°C, 5°C lower than the core body temperature. Loeb reported that the solubility of urate at 37°C and pH 7.4 is 404 μmol/L (6.8 mg/dl), while at 30°C it is 268 μmol/L (4.0 mg/dl). This means that if the uric acid concentration in the metatarsophalangeal joint cavity exceeds 268 μmol/L (4.5 mg/dl), crystal precipitation may occur. The typical foot arthritis in pain wind patients often occurs at night, possibly related to temperature reduction.
The self-termination of gouty arthritis can also be explained by temperature. Because during an acute attack, the local temperature rises, significantly increasing the solubility of sodium urate, and the microcrystals gradually dissolve and are absorbed, leading to the gradual subsidence of inflammation. Additionally, the body's stress state increases the secretion of adrenal cortical hormones and the excretion of sodium urate, which may also be another reason for the self-termination of acute attacks in patients.
3. Trauma and Other Influencing Factors: Hatz believes that mechanical injury to connective tissue is a triggering factor for attacks. Injury promotes the detachment of urate crystals from the synovial surface of the joint cavity, leading to pain wind attacks. Acute pain wind often occurs during outdoor camping and frequently affects the first metatarsophalangeal joint, which bears the most stress during walking.
Additionally, the blood supply to the joint cavity and surrounding tissues is relatively limited. During exercise, tissue oxygen consumption increases, and anaerobic glycolysis produces more lactic acid, leading to a decrease in pH, which can induce acute pain wind attacks.
(II) The Role of Leukocytes in the Attack Process
Polymorphonuclear leukocytes play an important role in the onset of acute arthritis caused by sodium urate microcrystals. Experiments have shown that after inducing leukopenia in animals with anti-leukocyte serum or vancomycin, sodium urate microcrystals cannot cause acute arthritis attacks. When leukocyte levels return to normal, inflammation attacks can often be induced. It is now understood that the generation and transformation reactions in the acute inflammatory process of pre-wind mainly include the following points:
1. Phagocytosis by polymorphonuclear leukocytes When sodium urate crystals on the surface of the synovial membrane in the joint cavity detach into the joint cavity, polymorphonuclear leukocytes in the synovial fluid and synovial membrane cells, mainly IgG immunoglobulins and other substances, adsorb and surround the microcrystals. The IgG-Fc segment can react with the Fc receptors of neutrophils, promoting the phagocytosis of the crystals by neutrophils. The phagocytosed sodium urate crystals can rapidly cause the dissolution of neutrophils, releasing lysosomal enzymes and enhancing the generation of superoxides in leukocytes.
2. Release of Chemokines Polymorphonuclear leukocytes phagocytose microcrystals, which are surrounded by a thin membrane to form phagosomes. The phagosomes fuse with primary lysosomes to form secondary lysosomes, which release leukocyte chemotactic factors C3a, C5a, and C567. These chemokines attract neutrophils to migrate towards the joint cavity.
3. Enzymatic Hydrolysis and Hydrogen Bonding After polymorphonuclear leukocytes phagocytose sodium urate crystals, phagosomes are formed. The interaction between phagosomes and lysosomes or the binding of hydrogen ions to the membrane of organelles rich in cholesterol and testosterone causes perforation of the organelles and rupture of the lysosomal membrane, releasing acid hydrolases and lysosomal enzymes. However, these enzymes cannot digest or hydrolyze sodium urate crystals, leading to the lysis and disintegration of leukocytes. The microcrystals, along with hydrolases and cytoplasmic enzymes released from the destroyed leukocytes, enter the surrounding tissues, causing inflammation. Subsequently, the microcrystals continue to be phagocytosed by other polymorphonuclear leukocytes, further exacerbating the inflammation.
bubble_chart Clinical Manifestations
Pain wind is typically divided into asymptomatic, acute, intermittent, and chronic stages. Its clinical manifestations have many characteristics, and familiarity with these characteristics enables clinical diagnosis for most patients. After the first episode of arthritis, pain wind may recur after weeks or even longer asymptomatic intervals. Subsequently, acute episodes become increasingly frequent in most patients. If left untreated, chronic sexually transmitted disease changes in tissues and organs such as joints and kidneys are inevitable.
I. Joint Lesions
(1) Acute Gouty Arthritis
Often onset is abrupt, with the first episode typically starting in the early morning, usually affecting only a single peripheral joint. About 50% of cases first involve the metatarsophalangeal joint of the big toe. Throughout the course of the disease, over 90% of patients experience involvement of this joint. Local joint pain, redness, and sometimes even shiny skin with visible venous dilation and ecchymosis, along with restricted movement, are common. Local symptoms worsen rapidly, peaking within hours, causing severe discomfort. Systemic symptoms such as malaise, aversion to cold, chills, and fever may accompany, with high fever reaching over 39℃, tachycardia, hepatomegaly, and marked polyuria. Mild cases may resolve spontaneously within hours or 1-2 days, while severe cases may last several days or weeks before subsiding. After inflammation subsides, the skin may appear dark red to purplish, wrinkled, with desquamation and grade I itching, gradually recovering.
Apart from the metatarsophalangeal joint, joints of the limbs can be affected, mostly the lower limbs, with more distal joint involvement showing more typical symptoms. The distribution and composition of joint involvement, as summarized from 879 domestic cases, are as follows: first metatarsophalangeal (58.7%), metatarsophalangeal (11.7%), metacarpophalangeal and interphalangeal (8.9%), ankle (8.7%), knee (3.9%), wrist (2.8%), with other joints being less common.
About 85% of acute episodes are triggered by: (1) heavy alcohol consumption or intake of purine-rich foods; (2) excessive fatigue or joint strain; (3) emotional stress or mental stimulation; (4) exposure to cold and damp; (5) surgery or trauma; (6) drug induction such as diuretics; (7) cancer chemotherapy or radiotherapy.
After acute gouty arthritis resolves, recurrence often occurs within a year. The frequency of recurrence varies widely, with 62% recurring within a year, 16% within 1-2 years, 11% within 2-5 years, 4% within 10 years, and 7% not recurring, based on an analysis of 1289 cases.
(2) Chronic Gouty Arthritis
With increasing frequency of acute episodes and disease progression, urate deposition in and around joints and other tissues gradually worsens, involving more joints and evolving into chronic arthritis, leading to joint deformity. The average time from initial onset to chronic arthritis formation is about ten years. A few cases may develop chronic sexually transmitted disease changes without acute episodes.
Due to urate deposition causing chronic inflammatory reactions, affected joints show asymmetric irregular swelling and progressive stiffness, leading to persistent pain, extensive destruction, and large subcutaneous nodules, ultimately resulting in joint deformity and loss of function.
Although chronic gouty arthritis can affect various joints and involve multiple joints simultaneously, it rarely affects spinal joints and costal cartilage, with mild symptoms if involved, sometimes presenting as chest pain, back pain, or intercostal neuralgia.
II. Pain Wind Nodules
Pain wind nodules, also known as pain wind stones, are caused by sodium urate deposition in tissues. Since urates do not easily cross the blood-brain barrier, pain wind nodules can form in almost all tissues except the central nervous system, most commonly in joint cartilage and surrounding tissues.
Typically, superficial pain wind nodules appear about ten years after onset. The incidence of pain wind nodules is related to serum urate levels as shown in Table 36-2.
| Urate concentration μmol/L(mg/dl) | Number of cases | Nodule size | Incidence rate % |
| <541.3(<9.1) | 722 | 0 | |
| 594.8~654.3(10~11) | 456 | Small to medium | 100 |
| >654.3(>11) | 111 | Large | 100 |
Some scholars have reported that when serum urate concentration is <535.4μmol/L(9mg/dl), about 5% of patients develop gouty tophi. Meng Zhaoheng et al. reported an incidence rate of 69.5% in 160 cases domestically, while Zhang Kaifu et al. reported 50.9% in 114 cases. Factors affecting the incidence rate of gouty tophi include: ① the level of serum uric acid; ② the duration of the disease; ③ the effectiveness of treatment.
The common sites for gouty tophi on the body surface are the external ear, especially the helix and antihelix; followed by the olecranon, knee joint capsule, and tendons; a few are seen on the fingers, palms, feet, eyelids, nasal cartilage, corneal membrane, or scleral membrane.
Characteristics of gouty tophi: ① protruding from the skin surface as yellowish or white round or oval nodules; ② varying in number from 1 to more than 10; ③ large ones are the size of an egg, small ones are only the size of a grain of rice; ④ texture is hard and tough or relatively soft; ⑤ as the volume increases, the epidermis becomes thin or injured and ulcerates, allowing white urate crystals to flow out.
III. Renal damage
Renal damage is the second most common clinical manifestation of gout, with about 20~40% of gout patients having renal lesions. The renal damage in gout is not related to the severity of gouty arthritis; grade I arthritis can have renal lesions, while severe arthritis patients may have no renal abnormalities. Common renal damages include the following.
(1) Urate nephropathy
Monosodium urate deposition in the renal medulla causes interstitial nephritis, leading to glomerular injury and eventually renal sclerosis. The initial manifestation is increased nocturia, decreased urine specific gravity, and mild to grade II proteinuria, initially intermittent, then progressing to persistent proteinuria. In addition, microscopic hematuria and increased white blood cells can be seen. The course is prolonged and slowly progressive; without treatment, azotemia occurs in 10~20 years. If accompanied by hypertension, pyelonephritis, diabetes, etc., it enters the uremic stage earlier. Some patients mainly have glomerular lesions, with a relatively rapid progression, leading to earlier renal failure.
(2) Acute uric acid nephropathy
In patients with severe hyperuricemia, a large amount of uric acid deposits in the collecting ducts in a short period, causing tubular obstruction, anuria, and acute renal failure. Before acute renal failure occurs, hematuric acid significantly increases, up to 4760μmol/L(80mg/dl), sand-like or stone-like urinary stones are seen in the urine, a large amount of uric acid crystals are found in the urine sediment, urine pH is significantly reduced, and the urine uric acid-creatinine ratio is >1.0.
(3) Uric acid urinary tract stones
The incidence of uric acid urinary stones in the normal population is 0.01%, while it is 20-25% in patients with primary pain wind and as high as 35-40% in secondary pain wind. The average age of stone occurrence is around 44 years old, and 40% of patients have urinary stones appearing before gouty arthritis, with 14% of these cases occurring more than 10 years prior. The composition of the stones is 84% pure uric acid rather than sodium urate, and 4% are mixed stones of uric acid and calcium oxalate. Pure uric acid stones are usually small, round, soft, brittle, yellow-red or brown, smooth, and dull. They are not visible on X-ray films, but if they are larger than 2.0 cm and impure in texture, faint shadows that are not transparent to light may be visible. They are more easily detected by contrast imaging. In some cases of uric acid stones, the main manifestations are renal colicky pain and microscopic hematuria, while some patients report cloudy crystalluria or the passage of sandy urine.
Patients presenting with the following conditions should be carefully evaluated for the possibility of stones: ① those with long-term urinary tract infections that come and go; ② those with long-term presence of small amounts of proteinuria (+ to ++) and small amounts of red and white blood cells in the urine, who do not respond to long-term treatment for nephritis; ③ those who seek medical attention for renal failure without a history of acute nephritis or acute pyelonephritis; ④ those with long-term acidic urine; ⑤ those with a family history of urinary tract stones. Stones can form at any stage of pain wind, and most patients do not have pain wind symptoms. Therefore, whenever encountering patients with urinary stones, it is necessary to carefully exclude pain wind and hyperuricemia.
IV. Cardiac Lesions
Urate can deposit in the inner membrane, outer membrane, valve membrane, myocardium, myocardial interstitium, and conduction system of the heart, and even form stones, causing myocardial damage, insufficient coronary blood supply, arrhythmia, and cardiac insufficiency. Some refer to this as "pain wind" heart disease. In the literature, there are reports of urate stones found in the mitral valve or cardiac conduction system, even causing complete atrioventricular block. However, it is rare for the cardiac manifestations of pain wind patients to be directly caused by urate; most are due to coexisting coronary heart disease.
V. Clinical Characteristics of Secondary Pain Wind
(1) Clinical Features of the Primary Disease
Any secondary hyperuricemia can cause secondary pain wind. The main diseases that can cause secondary hyperuricemia include those with increased nucleic acid metabolism and those with reduced renal excretion of urate (see Table 36-3). Among these, chronic myeloproliferative disorders and renal insufficiency caused by various diseases are the most common. Undoubtedly, these patients generally exhibit the clinical features of the primary disease. However, in a few cases, pain wind symptoms may appear before the symptoms of the primary disease, such as secondary pain wind symptoms caused by chronic myeloproliferative disorders, which can appear months or even years before the symptoms of the primary disease.
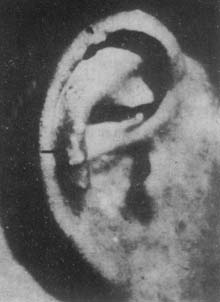
Table 36-3 Main Diseases Causing Secondary Pain Wind
| Increased Nucleic Acid Metabolism | Reduced Renal Excretion of Urate |
| Polycythemia Vera | Hypertensive Nephropathy |
| Secondary Polycythemia Due to Congenital Heart Disease or Chronic Lung Disease | |
| Chronic Myeloproliferative Disorders | Lead Poisoning Nephritis |
| Multiple Myeloma | Elevation of Plasma β-Hydroxybutyrate Due to Caloric Restriction |
| Chronic Hemolytic Anemia | Use of Drugs Such as Hydrochlorothiazide and Pyrazinamide |
| Acute or Chronic Leukemia | Interference with Urate Excretion |
| Lymphoma | |
| After Chemotherapy or Radiotherapy for Various Cancers |
(2) Serum Urate Levels Are Significantly Elevated
The serum urate levels in secondary pain wind are significantly higher than those in primary pain wind, with some reaching up to 4759 μmol/L (80 mg/dl). As seen in Table 36-4, 75% of secondary pain wind cases have levels >594 μmol/L, while only 31% of primary cases do. Concurrent with the increase in serum urate, the 24-hour uric acid excretion also significantly increases, as shown in Table 36-5. However, if the patient's renal excretion function is reduced, the 24-hour uric acid excretion may not increase.
(3) The symptoms of gout are atypical.
Due to the severe symptoms and short course of the primary disease, the symptoms of gouty arthritis are mild and atypical, rarely forming gout nodules. As a result, the symptoms of the primary disease often overshadow those of gout. Additionally, some primary diseases rapidly progress to a critical stage, making secondary gout easily overlooked.
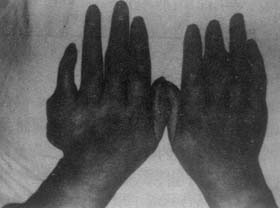
Table 36-4 Relationship between serum urate levels and the onset of gout
| Serum urate levels μmol/L (mg/dl) | Primary (%) | Secondary (%) |
| 297.4~351.0 (5.0~5.9) | 1 | 5 |
| 357.0~410.4 (6.0~6.9) | 2 | 0 |
| 416.4~470.0 (7.0~7.9) | 17 | 0 |
| 475.9~529.4 (8.0~8.9) | 26 | 10 |
| 535.4~588.9 (9.0~9.9) | 23 | 10 |
| 594.8~648.4 (10.0~10.9) | 17 | 15 |
| 654.3~707.9 (11.0~11.9) | 7 | 15 |
| 713.8~767.3 (12.0~12.9) | 4 | 15 |
| 773.3~1130.2 (13.0~19.0) | 3 | 30 |
Table 36-5 Relationship between 24-hour uric acid excretion and the onset of gout
| Uric acid excretion mmol/d (mg/24h) | Primary (%) | Secondary (%) |
| ≦2.36 (≦400) | 12 | 9 |
| 2.37~2.95 (401~500) | 17 | 3 |
| 2. 96~3. 54 (501~600) | 19 | 29 |
| 3.55~4.72(601~800) | 31 | 23 |
| 4.73~5.9(801~1000) | 16 | 17 |
| >5.9(>1000) | 6 | 20 |
(IV) Frequent kidney involvement
It goes without saying that primary diseases can lead to kidney lesions and induce renal insufficiency. Even secondary pain wind caused by increased nucleic acid metabolism can lead to oliguric or polyuric acute renal failure and the formation of urinary tract stones due to a significant increase in hematuria acid and massive excretion of uric acid.
bubble_chart Auxiliary Examination
Laboratory tests are of great significance for the diagnosis of pain wind, especially the discovery of urate, which is the basis for a definitive diagnosis.
I. Routine blood and urine tests and erythrocyte sedimentation rate (ESR)
1. Routine blood test and ESR examination During the acute stage of attack, the peripheral white blood cell count increases, usually (10-20)×109/L, rarely exceeding 20×109/L. The neutrophils increase accordingly. In patients with decreased renal function, mild to grade II anemia may occur. The ESR increases, usually less than 60mm/h.
2. Urine routine examination In the early stage of the disease, there is generally no change. In patients with kidney involvement, proteinuria, hematuria, and pyuria may occur, and occasionally casturia. In patients with concurrent kidney stones, obvious hematuria may be seen, and acidic urinary stones may also be discharged.
II. Hematuria acid measurement
During the acute stage of attack, the serum uric acid content increases in the vast majority of patients. It is generally believed that using the uricase method, a value of 416μmol/L (7mg/dl) for males and >357μmol/L (6mg/dl) for females has diagnostic value. If uric acid excretion drugs or adrenal corticosteroids have been used, the serum uric acid content may not be high. It can be normal during the stage of remission. About 2-3% of patients show typical pain wind attacks but have serum uric acid levels below the above levels. There are three explanations: ① The temperature gradient between the central body and the peripheral joints is large; ② The body is in a state of stress, secreting more adrenal corticosteroids, promoting serum uric acid excretion, while the sodium urate content in the distal joints remains relatively high; ③ The influence of using uric acid excretion drugs or corticosteroid treatment.
III. Urine uric acid content measurement
Under conditions of a purine-free diet and without taking drugs that affect uric acid excretion, the total 24-hour urine uric acid in normal adult males does not exceed 3.54mmol/(600mg/24h). 90% of primary pain wind patients excrete less than 3.54mmol/24h of urine uric acid. Therefore, normal urine uric acid excretion cannot rule out pain wind, while urine uric acid greater than 750mg/24h suggests excessive uric acid production, especially in non-renal secondary pain wind, where hematuria acid is elevated, and urine uric acid is also significantly elevated.
IV. Joint cavity puncture examination
During acute gouty arthritis attacks, there may be effusion in the swollen joint cavity. Extracting synovial fluid with an injection needle for examination has extremely important diagnostic significance. Even during the asymptomatic period, sodium urate crystals can be found in many joints. Urate crystals can be found in the synovial fluid of more than 95% of acute gouty arthritis cases. Figure 1 shows what is seen under the microscope.
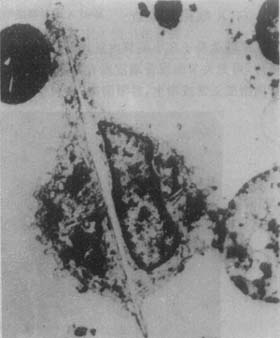
Figure 1 Electron micrograph of synovial fluid white blood cells phagocytosing sodium urate crystals
1. Polarized light microscopy
Place the synovial fluid on a glass slide, and slowly vibrating images of birefringent needle-shaped sodium urate crystals can be seen inside or outside the cells. Using a first-order red compensator, the urate crystals appear yellow when parallel to the microscope axis and blue when perpendicular.
2. Ordinary microscopy
Sodium urate crystals appear rod-shaped and needle-shaped, with a detection rate only half that of polarized light microscopy. If heparin is added to the synovial fluid, centrifuged, and the sediment is examined under the microscope, the detection rate can be improved.
3. UV spectrophotometry
Using UV spectrophotometry to qualitatively analyze the contents of the bursal fluid or suspected pain wind nodules to determine sodium urate is the most valuable method for pain wind. The method is to first measure the absorption spectrum of the sample to be tested, and then compare it with the known absorption spectrum of sodium urate. If they are the same, the measured substance is the known compound.
4. Murexide test
For specimens found to contain sodium urate upon examination with an ordinary optical microscope or polarizing microscope, this test can be performed for further confirmation, as it is simple and easy to conduct. The principle is that sodium urate, when heated with nitric acid, produces alloxan, which then reacts with ammonia solution to form ammonium purpurate, which appears purplish-red.
(5) Urate Dissolution Test
In synovial fluid containing urate crystals, after adding uricase and incubating, the urate crystals are degraded into allantoin, and the disappearance of crystals can be observed.
5. Examination of Pain Wind Nodule Contents
For pain wind nodules, biopsy or aspiration of their contents is performed, or chalky viscous material is collected from ulcers for smear examination. Using the above methods, the detection rate of specific urate crystals is extremely high.
6. X-ray Examination
Urate tends to deposit in and around small joints, causing chronic inflammatory reactions and destruction of cartilage and bone cortex. X-rays of these areas may show translucent defect shadows on the joint surface or bone cortex, appearing as punched-out, worm-eaten, honeycomb, or cystic patterns. The surrounding bone density is normal or hyperplastic, with clear boundaries, aiding in differentiation from other joint diseases. In a report by Meng Zhaoheng et al. on 160 cases, 76 cases (60.3%) showed the above typical changes in bone cortex, with 4 cases leading to fractures.
In summary, laboratory tests are indispensable for diagnosing pain wind and observing disease progression, especially the detection of urate crystals, which is key to improving the quality of pain wind diagnosis.
There is no unified standard for the diagnosis of pain wind in the country. Generally, the standards of the American Bi Disease Association, the Holmes criteria in the United States, and the revised criteria in Japan are often adopted. Here, we introduce the classification criteria for acute gouty arthritis by the American Bi Disease Association (1977):
1. Identification of specific urate crystals in synovial fluid;
2. Confirmation of sodium urate crystals in pain wind stones through chemical methods or polarized light microscopy;
3. Presence of 6 out of the following 12 clinical, laboratory, and X-ray signs.
⑴ More than one episode of acute arthritis;
⑵ Inflammation peaks within 1 day;
⑶ Monoarthritis attack;
⑷ Dark red skin over the affected joint;
⑸ Pain or swelling in the first metatarsus joint;
⑹ Unilateral attack involving the first metatarsus toe joint;
⑺ Unilateral attack involving the tarsal joint;
⑻ Suspected pain wind stones;
⑼ Hyperuricemia;
⑽ X-ray showing asymmetric joint swelling;
⑾ X-ray showing subcortical cysts without erosion;
⑿ Negative microbial culture of joint fluid during the attack stage of arthritis.
In summary, acute pain wind can be easily diagnosed based on typical clinical manifestations, laboratory tests, and treatment response. The diagnosis of chronic gouty arthritis requires careful differentiation and should ideally be based on the identification of urate crystals.
bubble_chart Treatment Measures
Dietary Therapy
Principles
(1) Maintain Ideal Body Weight
Epidemiological studies have found that serum urate levels are positively correlated with the degree of obesity, body surface area, and body mass index. Clinical observations indicate that when obese individuals lose weight, their serum urate levels decrease, urinary excretion decreases, and the frequency of gout attacks lessens.
(2) Limit Dietary Purine Intake
Some scholars recommend that daily purine intake should be kept below 100-150mg, especially by limiting the consumption of foods rich in purines.
Since protein has a special role in the body, excessive protein intake can also increase endogenous uric acid production, so it should also be appropriately limited.
(3) Encourage the Consumption of Alkaline Foods
Foods containing higher amounts of sodium, potassium, calcium, and magnesium, which oxidize in the body to form alkaline oxides, such as vegetables, potatoes, sweet potatoes, and dairy products, are physiologically referred to as alkaline foods. Fruits like citrus, after metabolism, leave behind rich alkaline elements like potassium, hence they are also considered alkaline foods. Increasing the intake of alkaline foods can reduce the acidity of serum and urine, even making the urine alkaline, thereby increasing the solubility of uric acid in urine.
(4) Ensure Adequate Urine Volume
If the patient's heart and lung functions are normal, maintaining a urine volume of about 2000ml/d is recommended to promote uric acid excretion. Therefore, the patient's total daily fluid intake should reach 2500-3000ml. Beverages should preferably include plain water, tea, mineral water, soda, and fruit juices. However, strong tea, coffee, cocoa, and similar beverages, which stimulate the autonomic nervous system, may trigger gout attacks and should be avoided. To prevent urine concentration at night, it is advisable to drink water before bed or during the night.
Methods
(1) Limit Total Caloric Intake
Total calories should be calculated based on the patient's ideal body weight under resting conditions, usually not exceeding 105-126kJ (25-30kcal)/kg per day. Clinical experience shows that adult patients with moderate to severe obesity (30-50% overweight) often cannot reduce their weight if their daily caloric intake exceeds 6300kJ. The following methods can be used to limit total caloric intake and aid in weight loss.
1. For patients 30-50% or more overweight, start with a total caloric intake of 6300kJ/d, divided into three meals. After one month, reduce to 5460kJ/d; or reduce caloric intake by 2310-4620kJ/d from the original diet, aiming for a weekly weight loss of 0.5-1.0kg.
2. For overweight or grade I obesity patients, start with a total caloric intake of 6300kJ/d, divided into three meals; or reduce caloric intake by 525-1050kJ/d from the original diet, aiming for a monthly weight loss of 0.5-1.0kg.
(2) Distribution of the Three Major Nutrients
Under the premise of limiting total caloric intake, the distribution of the three major nutrients should follow the principle of high carbohydrates, moderate protein, and low fat.
1. Carbohydrates, including vegetables and fruits, should account for 65-70% of total calories. This aligns with the dietary habits of the population and can reduce the production of ketones from fat breakdown, facilitating urate excretion. However, the intake of sucrose or beet sugar should be minimized.
2. Protein should account for 11-15% of total calories, typically 0.57-1.0g/kg per day. Preferred sources include milk, cheese, skim milk powder, and the protein part of eggs, as they are high-quality proteins rich in essential amino acids, necessary for tissue metabolism renewal, and contain very little purine, having almost no adverse effects on gout patients. However, yogurt, which contains more lactic acid, is unfavorable for gout patients and should not be consumed.
3. Fats The remaining portion of the total calories should be supplemented with fats, typically 40-50g/d. Since the oxidation of fats produces approximately twice as much heat as carbohydrates or proteins, it is undoubtedly necessary to limit them in order to reduce the patient's body weight.
Precautions
(1) Avoid Alcohol Consumption
The main component of alcohol is ethanol, which can induce glycogenolysis disorders, leading to the accumulation of lactic acid and ketone bodies in the body. The β-hydroxybutyrate in lactic acid and ketone bodies can competitively inhibit uric acid excretion.
(2) Limit Purine Intake Based on Individual Conditions
The restriction of purine intake should be tailored according to the severity of the patient's condition, the stage of the disease, complications, and the use of uric acid-lowering drugs, i.e., it should be in line with the individual's specific circumstances.
(3) Pay Attention to Food Cooking Methods
Proper cooking methods can reduce the purine content in food, such as boiling meat first and discarding the broth before further cooking. Additionally, foods and condiments like hot pepper, curry, pepper fruit, pricklyash peel, mustard, and fresh ginger rhizome can stimulate the autonomic nervous system and trigger acute attacks of gout, so their use should be minimized.
Clinical Pharmacology of Anti-Gout Drugs
Currently, there is no cure for gout. The goals of drug treatment are limited to: ① Quickly terminating acute attacks and preventing the recurrence of acute arthritis; ② Preventing and treating the deposition of urate in joints, kidneys, and other tissues; ③ Preventing uric acid kidney stones; ④ Treating complications such as hypertension, hyperlipidemia, and diabetes.
Anti-gout drugs can be summarized into the following six categories (Table 36-9).
Table 36-9 Classification of Anti-Gout Drugs
| Gout Inflammation Interference Drugs | Uric Acid-Lowering Drugs |
| (1) Colchicine | (1) Uricosuric Drugs |
| (2) Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) | (2) Uric Acid Synthesis Inhibitors |
| (3) Adrenocorticosteroids | (3) Drugs with Dual Pharmacological Effects |
| 1. Uricosuric Drugs with Hypoglycemic Effects 2. Uricosuric Drugs with Lipid-Lowering Effects 3. Uricosuric Drugs with Antihypertensive Effects |
I. Gout Inflammation Interference Drugs
(1) Colchicine
Since the 6th century, this drug has been used to treat acute gout. It has selective anti-inflammatory effects on gout and can interfere with the inflammatory response to urate microcrystals. Therefore, it remains the first-line treatment for gout, especially for severe acute attacks, and can relieve pain and inflammation in over 90% of patients within 12 hours, with symptoms disappearing within 24-48 hours. However, local swelling may persist for several days or longer. Its possible mechanisms of action include: ① Inhibiting the chemotaxis, proliferation, and phagocytosis of polymorphonuclear leukocytes; ② Inhibiting the release of lysosomes and lactic acid; ③ Increasing the pH in the joint cavity, reducing the precipitation of urate crystals. However, it does not lower blood uric acid levels nor increase uric acid excretion.
Usage: Oral administration, initial dose of 0.5~1.0mg, followed by 0.5mg every hour until pain relief or severe gastrointestinal reactions become intolerable, then switch to a maintenance dose of 0.5mg, 1~3 times daily. Generally, taking 5mg within 10~12 hours results in minimal gastrointestinal reactions and excellent efficacy. The maximum tolerable dose should not exceed 6~8mg. Intravenous administration offers the advantages of rapid effect and fewer gastrointestinal reactions, making it particularly suitable for acute attacks in ulcer disease or during the convalescence stage post-surgery. The method involves dissolving 2mg in 20ml of saline for slow intravenous injection, and depending on the condition, an additional 1mg can be administered after 4~6 hours, but the total dose during one episode should not exceed 4~5mg. For those already on preventive medication, the total dose should not exceed 2mg. It is noteworthy that intravenous administration results in fewer gastrointestinal reactions, making toxicity difficult to detect, thus blood leukocyte counts should be checked before and after administration. This drug has strong local irritant effects, hence it must not be administered outside the blood vessels in fistula disease.
Side effects and their management: Gastrointestinal reactions such as abdominal pain, nausea, vomiting, and diarrhea often occur when symptoms are relieved. In severe cases, hemorrhagic gastroenteritis may occur. A few cases may cause leukopenia, aplastic anemia, alopecia areata, and myopathy after medication. If diarrhea occurs and medication needs to be continued, Imodium can be taken or 1-4ml of compound formula camphor tincture can be taken after each bowel movement until diarrhea stops. Long-term medication must monitor blood counts, and those with low bone marrow function should avoid use. Dosage should be appropriately reduced for those with liver and kidney diseases. This drug can cause birth defects and should be completely avoided during the first three months of pregnancy. Additionally, it can enhance the effects of sedatives, hypnotics, analgesics, and anesthetics; it can also enhance the effects of amphetamines, adrenaline, and ephedrine; it reduces the effects of anticoagulants and antihypertensive drugs, so attention should be paid to drug interactions when used in combination, and dosage should be adjusted accordingly.
(II) Non-steroidal anti-inflammatory drugs
Non-steroidal anti-inflammatory drugs (NSAIDs) are very effective in treating most acute pain wind, and the side effects of these drugs are smaller than those of colchicine, and they are effective even when administered several days after the onset of an attack. Additionally, the efficacy of NSAIDs is also limited by side effects. The most common side effects are gastrointestinal reactions and kidney damage. The former includes indigestion, nausea, upper abdominal pain, ulcers, bleeding, etc.; the latter includes nephrotic syndrome, interstitial nephritis, renal papillary necrosis, and acute kidney failure. NSAIDs inhibit cyclooxygenase, which is generally not very important for patients with normal renal function, but in patients with renal insufficiency or heart failure, treatment with these drugs may exacerbate hypertension, water retention, and azotemia.
Indomethacin (Indocin) is used to treat acute attacks of pain wind, usually 5mg, 3-4 times a day until symptoms significantly improve, then reduced to 25mg, 3-4 times a day. It can relieve arthralgia in 90% of patients within 2-4 hours, but has the following main side effects: ① gastrointestinal reactions, which can cause ulcer disease or gastrointestinal bleeding in severe cases; ② headache, vertigo; ③ occasionally causes rash, asthma, leukopenia, temporary depersonalization, etc.
Other NSAIDs such as oxyphenbutazone, ibuprofen, naproxen, piroxicam, ketoprofen, meclofenamic acid, etc. have been proven effective in treating acute pain wind. Full therapeutic doses should be used initially until clinical symptoms significantly improve, then reduced until completely discontinued. The choice and use of drugs can be determined based on the patient's condition and the toxic side effects of the drugs. The pharmacokinetics, clinical characteristics, and usage of commonly used NSAIDs.
(III) Adrenocorticosteroids
In cases of severe acute gout attacks accompanied by significant systemic symptoms, when colchicine or NSAIDs are ineffective, intolerable, or contraindicated, the use of this class of drugs may be considered. Among them, adrenocorticotropic hormone (ACTH) is the most effective. Commonly, 25-50U is added to 500ml of glucose solution for intravenous drip, maintained over 8 hours, once daily, or 50U intramuscularly every 6-8 hours, both for 2-3 consecutive days. Alternatively, hydrocortisone succinate 200-300mg can be administered intravenously once daily, or prednisone 30mg/day, taken orally in divided doses. Due to the rebound phenomenon after withdrawal of ACTH or corticosteroids, it is best to concurrently and subsequently use maintenance doses of colchicine or indomethacin for one week. For lesions confined to individual joints, hydrocortisone acetate 25-50mg can be injected locally into the joint cavity. Alternatively, triamcinolone 10-25mg, prednisolone acetate 25mg, or hydrocortisone diacetate 5mg can be used for local injection, with pain often completely relieved within 12-24 hours. The efficacy of using this class of drugs combined with anesthetics for intra-articular injection is increasingly recognized, such as triamcinolone 5-20mg plus 2% lidocaine 2-3ml, or 0.25% procaine 10-20ml, or 0.75% bupivacaine 2-3ml, with the latter maintaining a longer duration.
II. Uric Acid-Lowering Drugs
Uric acid-lowering drugs generally do not have anti-inflammatory or analgesic effects. Due to the rapid decrease in serum uric acid levels in patients, they can trigger acute attacks of gout (uric acid transfer attacks) or delay the relief of acute attacks. Therefore, when starting uric acid-lowering drugs, prophylactic anti-inflammatory drugs for gout should be administered until the serum uric acid level drops below 375 μmol/L (6 mg/dl).
(1) Uricosuric Agents
This class of drugs has the following three effects: ① Inhibit the reabsorption of uric acid in the renal tubules; ② Increase the secretion of uric acid in the renal tubules; ③ Increase the glomerular filtration rate of uric acid. The primary effect is to inhibit the reabsorption of uric acid and increase its excretion. To prevent the formation of uric acid stones in the urinary tract, urine should be alkalized during medication, maintaining a morning urine pH of 6.2–6.5, and ensuring adequate urine volume.
1. Probenecid (Probenecidum) The first uricosuric agent discovered in 1950. It is completely absorbed in the gastrointestinal tract, with a serum half-life of 6–12 hours, and 70% is eliminated from circulation within 24 hours, but its metabolites still have uricosuric effects. Therefore, its maximum therapeutic effect occurs several days after administration. A daily dose of 0.5 g can increase urinary uric acid excretion by 24–45%; if 2 g is taken daily, it increases by 60%. Generally, start with 0.25 g twice daily. Increase by 0.5 g weekly until serum uric acid levels return to normal, but the maximum daily dose should not exceed 3 g.
Main side effects: gastrointestinal reactions, fever, rash, etc., and occasionally hemolytic anemia. Since this drug belongs to the sulfonamide class, it is contraindicated in patients allergic to sulfonamides.
2. Sulfinpyrazone This drug is a derivative of phenylbutazone and therefore has weak anti-inflammatory and analgesic effects. Its uricosuric effect is significantly stronger than probenecid; a daily dose of 300–400 mg is equivalent to 1.0–1.5 g of probenecid. It is well absorbed in the gastrointestinal tract, and a single dose can last for 10 hours. This drug also inhibits platelet aggregation and prolongs platelet survival time, making it particularly suitable for patients with hemodynamic changes. The initial dose is 50 mg twice daily, increasing by 100 mg weekly until serum uric acid levels return to normal. However, the maximum daily dose should not exceed 800 mg. The side effects and contraindications of this drug are the same as those of phenylbutazone. Literature reports 10–15% of patients experience gastrointestinal reactions, and个别 cases of renal failure have been reported during use.
3. Benzbromarone (Benzbromarone, Gout, etc.) This drug is a benzofuran derivative. It is easily absorbed orally, and serum uric acid levels begin to decrease within 3 hours after administration, reaching peak uric acid clearance 4–5 hours later. Serum uric acid levels decrease by 66.5% after 24 hours, and the effect lasts for 48 hours. It is particularly suitable for patients who cannot use probenecid or allopurinol or those with extensive gouty tophi. The dosage is 40–80 mg (microcrystalline tablets) or 100–200 mg (non-microcrystalline tablets) taken every morning. Main side effects include gastrointestinal disturbances, renal colic, acute gout attacks, rash, etc., and occasionally bone marrow suppression. It is ineffective in patients with a glomerular filtration rate below 20 mg/min. The overall efficacy rate of this drug in treating gout in China is 89%.
(2) Uric Acid Synthesis Inhibitors
The prominent representative of this class of drugs is allopurinol (alopurinol), whose chemical structure is similar to hypoxanthine.
By competitively inhibiting xanthine oxidase, it prevents hypoxanthine from being oxidized to xanthine, and xanthine from being converted to uric acid. The human kidney has a higher clearance rate for hypoxanthine and xanthine than for uric acid, and hypoxanthine is highly soluble, so it does not cause damage to the kidneys. After absorption, it is metabolized by the liver into isoxanthine, which is easily soluble in water, and is excreted in urine. The half-life of this drug is 1-3 hours. Serum uric acid begins to decrease 1-2 days after taking the drug, peaks at 7-14 days, and usually returns to normal within 3-6 months. The indications for this drug include: ① Patients whose 24-hour uric acid excretion remains greater than 600mg (3054mmol) after low-purine diet treatment; ② Patients who are unresponsive, allergic, or intolerant to uric acid excretion drugs; ③ Patients with significantly reduced renal function and uric acid nephropathy or uric acid urinary stones; ④ Before starting chemotherapy or radiotherapy for lymphoproliferative or granulocytic proliferative diseases; ⑤ Severe gout with a large accumulation of urate, hyperuricemia, or no increase in uric acid excretion, and no urinary stones.
Start with an oral dose of 50mg, 2-3 times daily, then increase by 100mg weekly or biweekly. The maximum dose for severe cases is 1000mg/day. The commonly used dose in the country is 300-600mg/day. After 1-3 weeks, serum uric acid levels drop to 178.4-297.4μmol/L (3-5mg/dl), blood urea nitrogen decreases, and creatinine clearance returns to normal. The maintenance dose depends on serum uric acid levels, usually 0.1-0.2g, 2-3 times daily. If combined with uric acid excretion drugs, the dose should be increased appropriately because uric acid excretion drugs can increase the excretion of active metabolites of allopurinol.
The incidence of side effects is about 3-5%. Common side effects include: ① allergic rash, urticaria, drug fever, eosinophilia, etc.; ② bone marrow suppression leukopenia, hemolytic anemia; ③ toxic hepatitis or transient elevation of alanine aminotransferase; ④ vasculitis and eye damage; ⑤ xanthine stones. The author has observed two cases of severe exfoliative dermatitis in the treatment of hyperuricemia with this drug, both of which were cured by discontinuing the drug and applying corticosteroids.
(III)Dual pharmacological action category
1. Uric acid excretion drugs with hypoglycemic effect. Halofenate has a chemical structure similar to clofibrate (etofylline clofibrate), both being derivatives of phenoxyacetic acid. After oral administration, it is rapidly hydrolyzed to the free acid form and extensively bound to plasma proteins, with about 50% excreted in urine. The plasma half-life is 43h, significantly longer than clofibrate. The active components secreted in urine can be inhibited by probenecid and para-aminohippuric acid (PAH). When the urine is acidic, it is not affected by the diuretic effect of mannitol, but when it is alkaline, secretion increases and is further enhanced by the addition of diuretics.
The lipid-lowering mechanism of halofenate is to inhibit liver lipid synthesis, with a stronger effect on triglycerides than on cholesterol, reducing triglycerides by an average of 45-50%. The uric acid excretion effect is partly due to the inhibition of renal tubular reabsorption of uric acid. Its efficacy is similar to probenecid, and oral administration of 0.25g three times daily can reduce serum urate and increase urinary uric acid. Additionally, it has the effects of inhibiting platelet aggregation and lowering blood sugar. Since pain wind patients often have type IV hyperlipoproteinemia, hyperglycemia, and increased platelet aggregation, it is a valuable drug for the treatment of pain wind. The main side effects are stomach discomfort and rash.
2. Uric acid excretion drugs with lipid-lowering effect. Acetohexamide is a sulfonylurea oral hypoglycemic drug. It was accidentally discovered to have a significant uric acid excretion effect while lowering blood sugar. Its hypoglycemic mechanism is the same as other sulfonylurea hypoglycemic drugs, mainly stimulating pancreatic β cells to release insulin, and secondarily reducing liver glycogen release and blocking liver insulinase action. The uric acid promotion mechanism is mainly due to its side chain cyclohexyl group, which inhibits renal tubular reabsorption of uric acid.
Unlike most sulfonylurea hypoglycemic drugs, this drug is easily decomposed into hydroxyhexamide, with a biological half-life of about 1.3h; but hydroxyhexamide can enhance the hypoglycemic effect by 2.5 times, extending the effect for 12-24h; the uric acid excretion effect lasts for 8-10h. The daily dose is 500-1500mg, taken once or divided into two doses. For patients with renal insufficiency, the dual pharmacological effects are separated, meaning only the hypoglycemic effect is present, without the uric acid excretion effect.
3. Uric acid excretion drugs with antihypertensive effect. Tinilie acid and indanyloxyacetic acid are derivatives of ethacrynic acid. However, their drug effects are significantly different from ethacrynic acid. The former two have both uric acid excretion and antihypertensive effects, while the latter increases serum uric acid while diuresis, so it is not suitable for pain wind patients.
The mechanism of uric acid excretion promotion is to inhibit the reabsorption of uric acid in the proximal convoluted tubule; the blood pressure-lowering mechanism is the same as that of thiazide diuretics, which is to block the reabsorption of sodium and water in the early distal convoluted tubule, thereby reducing blood volume, decreasing cardiac output, and lowering peripheral stirred pulse resistance.
Management of Pain Wind at Different Stages
To facilitate timely and appropriate clinical management for specific patients, the key points of management based on the different clinical stages of Pain Wind are briefly described below:
1. Management of Asymptomatic Hyperuricemia
Hyperuricemia is defined as serum uric acid levels greater than 417 μmol/L in men and 357 μmol/L in women, commonly seen in individuals with a family history of Pain Wind, with an incidence rate of 1.4% in the urban adult population. The primary management during this stage involves dietary therapy and avoiding triggering factors such as alcohol abuse, while actively treating obesity, hypertension, hyperlipidemia, diabetes, etc.
Currently, most scholars believe that about 80% of asymptomatic hyperuricemia cases do not develop symptoms throughout life, and only a small portion may develop conditions like gouty arthritis or kidney damage after many years. The risk of uric acid kidney stones is related to increased urinary uric acid excretion rather than closely related to serum uric acid levels. However, drug treatment should be considered under the following conditions: serum uric acid levels greater than 535.5 μmol/L; family history of Pain Wind or urinary uric acid excretion greater than 5.9 mmol/d, or urinary uric acid greater than 4.1 mmol/d on a low-purine diet, with potential for uric acid kidney stones or acute uric acid nephropathy.
2. Management of Acute Pain Wind
After a prolonged period of asymptomatic hyperuricemia, acute gouty arthritis can suddenly occur. At this time, prompt and appropriate management can often achieve significant effects in terminating the attack.
Many authors have repeatedly emphasized the following characteristics for this stage of treatment: ① Early administration of medication is crucial. Pain Wind inflammation-interfering drugs should be given as soon as signs of an acute attack appear. Small doses can often control acute attacks, with outpatient patients commonly given ibuprofen, naproxen, etc., while hospitalized patients are primarily treated with colchicine; ② Treatment to control acute attacks should continue until inflammation completely subsides. Premature discontinuation or resumption of physical activity often leads to recurrence; ③ Uric acid excretion drugs and production inhibitors can prolong the acute attack process. Uric acid-lowering drugs should not be used alone during this stage; ④ Drugs affecting serum uric acid excretion, such as penicillin, thiazides, furosemide, vitamin B1, B2, insulin, ethambutol, pyrazinamide, levodopa, etc., should be avoided; ⑤ Properly manage triggering factors such as acute infections, surgical procedures, acute blood loss, and mental stress; ⑥ Do not neglect rest, dieta


