| disease | Dilated Cardiomyopathy |
The characteristic features of this type include dilation of the left or right ventricle or both ventricles, accompanied by myocardial hypertrophy. Ventricular systolic function is impaired, with or without congestive heart failure. Ventricular or atrial arrhythmias are common. The condition progresses progressively, and death can occur at any stage of the disease.
bubble_chart Etiology
The disease cause of this illness remains unclear to date, but the following factors have been identified as potentially related:
1. **Infection** In animal experiments, Coxsackievirus and encephalomyocarditis virus can not only cause viral myocarditis but also lead to lesions similar to dilated cardiomyopathy. Clinical follow-ups of patients with acute sexually transmitted disease viral myocarditis have shown a significantly higher likelihood of progression to dilated cardiomyopathy compared to the general population. Inflammatory manifestations have been observed in myocardial biopsy samples from these patients, and many exhibit higher titers of Coxsackievirus B neutralizing antibodies in their blood than normal individuals. Recent molecular biology techniques have detected the RNA of enteroviruses or cytomegaloviruses in myocardial biopsy samples from these patients, further supporting a close relationship between this disease and viral myocarditis. It is possible that this disease results from persistent infection.
2. **Genetics and Autoimmunity** Research has found that this disease is associated with histocompatibility antigens. Compared to non-affected individuals, patients with this disease show increased frequencies of HLA B27, HLA A2
, HLA DR4, and HLA DQ4, while the HLA DRw6 locus is decreased. These HLA variations are linked to autosomal recessive inheritance, which may explain the familial tendency observed in some patients. Additionally, altered immune responses may increase susceptibility to viral infections, leading to cardiac autoimmune injury.3. **Cellular Immunity** In patients with this disease, natural killer cell activity is reduced, weakening the body's defense mechanisms. The number and function of suppressor T lymphocytes are also diminished, resulting in cell-mediated immune responses that cause vascular and myocardial injury.
In summary, the current hypothesis regarding the possible mechanism of disease is as follows: Initially, Coxsackievirus invades the myocardium, proliferates, and causes myocardial cell necrosis. In the second stage, the virus is no longer detectable in the myocardium, but lymphocyte infiltration increases. These sensitized lymphocytes trigger immune responses, leading to further myocardial cell necrosis. In the late stage (third stage), inflammatory cell infiltration decreases or disappears, replaced by fibrosis, which interweaves with hypertrophied or reduced myocardial cells, forming the pathological changes characteristic of dilated cardiomyopathy. Although the viral infection and immune response theories are currently the leading explanations for the disease's pathogenesis, many questions remain unanswered, requiring further research.
bubble_chart Pathological Changes
The heart weight increases, approximately double the normal weight. All cardiac chambers are enlarged, with the myocardium appearing pale and flabby. Although the ventricular wall myocardium is hypertrophied, the wall thickness remains nearly normal due to chamber dilation. The endocardium may also thicken. Mural thrombosis within the cardiac chambers is not uncommon. Myocardial fibrosis is common, appearing as focal distributions along the inner edges of the ventricular walls, or sometimes as extensive damage to the heart wall. The cardiac pacemaker and conduction system can also be affected.

Electron microscopy reveals swollen mitochondria in myocardial cells, with cristae fragmentation or disappearance; widened gaps between the sarcoplasmic membranes, containing fibrous material and granular lipofuscin; and possible disappearance of myofibrils.
The myocardial lesions weaken cardiac contractility. In the early stage, the rate of left ventricular pressure rise during isovolumic contraction slows, and ejection velocity also decreases. At this stage, the reduced stroke volume is compensated by an accelerated heart rate, maintaining cardiac output. Later, incomplete left ventricular emptying leads to residual blood volume, increased end-diastolic pressure, and progressive development of left heart failure. Over time, left atrial and pulmonary artery pressures rise successively, eventually resulting in right heart failure. In a few cases where the lesions primarily affect the right ventricle, right heart failure develops directly. Ventricular dilation enlarges the atrioventricular valve rings, causing mitral or tricuspid regurgitation. Chamber dilation increases ventricular wall tension and oxygen consumption, while myocardial hypertrophy and accelerated heart rate cause relative myocardial ischemia. Since myocardial oxygen extraction has already reached its limit, this can lead to cardiac colicky pain. Involvement of the pacemaker and conduction system by myocardial lesions can cause various arrhythmias.

The disease can occur at any age, but is most common in middle-aged individuals. The onset is usually slow, with initial examinations revealing an enlarged heart and compensated cardiac function without noticeable discomfort. Symptoms gradually appear after a period of time, sometimes taking over 10 years to develop. The primary symptoms are those of congestive heart failure, with shortness of breath and edema being the most common. Initially, shortness of breath occurs after exertion or fatigue, but later it may appear during grade I activity or even at rest, or manifest as paroxysmal nocturnal dyspnea. Due to low cardiac output, patients often experience weakness. Physical examination reveals an accelerated heart rate, the apex beat displaced downward and to the left, possibly with a heaving impulse, the cardiac dullness border expanded to the left, and often audible third or fourth heart sounds, which may form a gallop rhythm when the heart rate is rapid. Due to cardiac chamber dilation, systolic blowing murmurs caused by relative mitral or tricuspid insufficiency may be heard, and these murmurs diminish as cardiac function improves. Blood pressure is mostly normal, but in advanced cases, it may decrease with a narrowed pulse pressure. During heart failure, diastolic pressure may rise to grade I. The appearance of pulsus alternans suggests left heart failure. The pulse is often weak. During heart failure, rales may be heard at the lung bases. In right heart failure, the liver becomes enlarged, and edema typically starts in the lower limbs. Pleural effusion and ascites are common in advanced cases. Various arrhythmias may occur, either as the first or primary manifestation, and multiple arrhythmias may coexist, forming complex rhythms that can recur and sometimes prove stubborn. High-grade atrioventricular block, ventricular fibrillation, sinoatrial block, or pauses can lead to Adams-Stokes syndrome, which may be fatal. Additionally, embolisms in the brain, kidneys, lungs, or other organs may occur.
bubble_chart Auxiliary Examination
X-ray examination shows an enlarged cardiac shadow, with an advanced stage appearance resembling a spherical shape, indicating enlargement of all cardiac chambers, similar in appearance to pericardial effusion. In a few patients, the enlargement is predominantly in the left ventricle, left atrium, or right ventricle, resembling mitral valve disease. Fluoroscopy reveals weaker cardiac pulsations compared to normal. The main pulmonary artery is generally not enlarged. Patients with a longer disease course often exhibit pulmonary congestion and interstitial edema, with septal lines visible at the costophrenic angles of both lungs. The pulmonary veins and pulmonary artery shadows may be enlarged; pleural effusion is not uncommon.
Electrocardiogram (ECG) findings are almost always abnormal in symptomatic patients, and many asymptomatic individuals already show ECG changes. These changes primarily include cardiac hypertrophy, myocardial damage, and arrhythmias. Left ventricular hypertrophy is common, often accompanied by myocardial strain, and advanced stages frequently show right ventricular hypertrophy. Left or right atrial hypertrophy may also occur. Myocardial damage is common, mainly manifesting as ST-segment depression, flattened or biphasic or inverted T waves, and sometimes ischemic T-wave changes. A few patients may exhibit pathological Q waves, resembling myocardial infarction, typically in the anterior septum (leads V1 and V2), possibly due to septal fibrosis. Intraventricular conduction blocks are common, including left or right bundle branch blocks or fascicular blocks. Arrhythmias are frequent, especially in late-stage [third-stage] disease, predominantly ectopic rhythms and conduction blocks. Ectopic rhythms may originate from the atria, atrioventricular junction, or ventricles, progressing from premature beats to tachycardia, flutter, or fibrillation. Sinus node dysfunction, atrioventricular junctional escape rhythms, or ventricular rhythms may also occur. First- to third-degree atrioventricular blocks can develop (Figure 1).
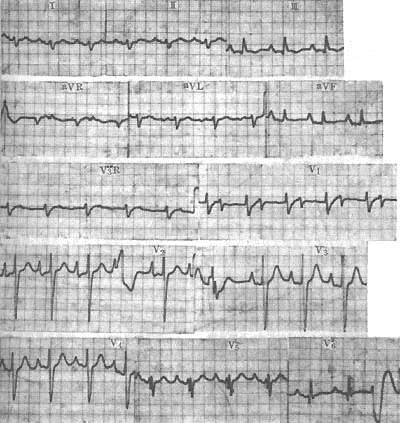
Figure 1 ECG of dilated primary cardiomyopathy
The ECG shows atrial hypertrophy, left posterior fascicular block, first-degree atrioventricular block, and ventricular premature beats. The diagnosis was confirmed by autopsy.
Echocardiography can detect early-stage cardiac chamber enlargement, particularly in the left ventricle, with weakened wall motion. In late-stage [third-stage] disease, all cardiac chambers are enlarged, and the motion of the interventricular septum and left ventricular posterior wall is also weakened. The double-peak appearance of the anterior mitral valve leaflet may disappear, and the anterior and posterior leaflets may exhibit paradoxical motion. The left ventricular ejection fraction often decreases below 50%, and the myocardial shortening fraction is also reduced. A small amount of pericardial effusion may be present.
Radionuclide ventriculography can also reveal cardiac chamber enlargement and weakened wall motion, with a reduced left ventricular ejection fraction, which becomes more pronounced after exercise.
Systolic time intervals may be abnormal early on, with a shortened left ventricular ejection time (LVET), prolonged pre-ejection period (PEP), and an increased PEP/LVET ratio.
Cardiac catheterization findings are nearly normal in the early stages, with slightly elevated left and right ventricular end-diastolic pressures. In heart failure, the cardiac index decreases, the arteriovenous oxygen difference widens, and pulmonary artery and atrial pressures rise. Cardiac angiography shows enlarged cardiac chambers and weakened wall motion.
In 1980, the World Health Organization stated that this disease is characterized by unexplained enlargement of the left ventricle or both ventricles, impaired ventricular systolic function, with or without congestive heart failure and arrhythmias. The diagnosis can only be made after excluding other causes.
In 1995, the Chinese Heart and Blood Vessel Society organized a symposium and proposed the following diagnostic criteria for this disease:
1. Clinical manifestations include cardiac enlargement, reduced ventricular systolic function with or without congestive heart failure, often accompanied by arrhythmias, and complications such as embolism and sudden death.
2. Cardiac enlargement: Chest X-ray shows a cardiothoracic ratio >0.5. Echocardiography reveals generalized cardiac enlargement, particularly significant left ventricular enlargement, with a left ventricular end-diastolic diameter ≥2.7 cm/m2, and the heart may appear spherical.
3. Reduced ventricular systolic function: Echocardiography shows diffuse weakening of wall motion and an ejection fraction below the normal value.
4. Other specific (secondary) cardiomyopathies and endemic cardiomyopathies (such as Keshan disease) must be excluded, including ischemic cardiomyopathy, peripartum cardiomyopathy, alcoholic cardiomyopathy, cardiomyopathies caused by metabolic and endocrine disorders (e.g., hyperthyroidism, hypothyroidism, amyloidosis, diabetes), cardiomyopathies due to hereditary familial neuromuscular disorders, cardiomyopathies caused by systemic diseases (e.g., systemic lupus erythematosus, rheumatoid arthritis), and toxic cardiomyopathies. Only then can idiopathic dilated cardiomyopathy be diagnosed.
If possible, serum antibodies against myocardial peptides such as anti-mitochondrial ADP/ATP carrier antibodies, anti-myosin antibodies, anti-β1-receptor antibodies, and anti-M2 cholinergic receptor antibodies can be detected as auxiliary diagnostic markers. For cases difficult to distinguish from coronary heart disease clinically, coronary angiography is required.
Endomyocardial biopsy: Pathological examination is not specific for the diagnosis of this disease but helps differentiate it from specific cardiomyopathies and acute myocarditis. Polymerase chain reaction (PCR) or in situ hybridization on endomyocardial biopsy specimens can aid in the diagnosis of infectious causes or perform genetic analysis for specific cellular abnormalities.
(1) Rheumatic heart disease: Cardiomyopathy may also present with systolic murmurs in the mitral or tricuspid areas, but generally without diastolic murmurs. The murmurs are louder during heart failure and diminish or disappear after heart failure is controlled, which contrasts with rheumatic heart disease. Cardiomyopathy often involves simultaneous enlargement of multiple cardiac chambers, unlike rheumatic heart disease, which primarily affects the left atrium, left ventricle, or right ventricle. Echocardiography helps differentiate the two.
(2) Pericardial effusion: Cardiomyopathy with cardiac enlargement and weakened heartbeats must be distinguished from pericardial effusion. In cardiomyopathy, the apical impulse is displaced downward and to the left, aligning with the left outer border of cardiac dullness. In pericardial effusion, the apical impulse is often indistinct or located medial to the left outer border of cardiac dullness. Systolic murmurs in the mitral or tricuspid areas, ventricular hypertrophy on electrocardiogram, abnormal Q waves, and various complex arrhythmias all indicate cardiomyopathy. Echocardiography easily differentiates the two: a large amount of fluid or anechoic space in the pericardium suggests pericardial effusion, while cardiac enlargement indicates cardiomyopathy. It should be noted that cardiomyopathy may also have a small amount of pericardial effusion, but this is insufficient to cause cardiac tamponade or affect cardiac signs and function, being only an incidental finding on ultrasound. Systolic time intervals are markedly abnormal in cardiomyopathy but normal in pericardial disease.
(3) Hypertensive heart disease: Cardiomyopathy may present with transient hypertension, but the diastolic pressure usually does not exceed 14.67 kPa (110 mmHg) and occurs during acute heart failure, decreasing after heart failure improves. Unlike hypertensive heart disease, fundus examination, urinalysis, and renal function are normal.
(4) Coronary Heart Disease For middle-aged and older patients, if there is cardiac enlargement, arrhythmia, or heart failure without other identifiable causes, coronary heart disease and cardiomyopathy must be considered. The presence of predisposing factors such as hypertension, hyperlipidemia, or diabetes, along with segmental abnormalities in ventricular wall motion, supports the diagnosis of coronary heart disease. In recent years, the condition where long-term, widespread myocardial ischemia due to coronary artery disease leads to fibrosis and progresses to cardiac dysfunction has been termed "ischemic cardiomyopathy." If there is no history of angina or myocardial infarction, it can be difficult to distinguish from cardiomyopathy. Additionally, cardiomyopathy may also present with pathological Q waves and angina. In such cases, differentiation relies on coronary angiography.
(5) Congenital heart disease Most have obvious signs and are not difficult to distinguish. Ebstein's anomaly is characterized by a murmur in the tricuspid valve area, and may also present with gallop rhythm, weakened cardiac impulse, right heart enlargement, and failure, which must be differentiated from cardiomyopathy. However, this condition manifests symptoms early in life, with no left ventricular enlargement and more pronounced cyanosis. Echocardiography can confirm the diagnosis.
(6) Secondary cardiomyopathy Systemic diseases such as systemic lupus erythematosus, scleroderma, hemochromatosis, amyloidosis, glycogen storage disease, and neuromuscular disorders all have manifestations of their primary diseases for differentiation. More importantly, distinguishing it from myocarditis is crucial. Acute myocarditis often occurs during or shortly after a viral infection, making differentiation relatively straightforward. Chronic myocarditis without a clear history of acute myocarditis is difficult to distinguish from cardiomyopathy. In fact, many cases of dilated cardiomyopathy develop from myocarditis, referred to as "post-myocarditis cardiomyopathy."
In recent years, endomyocardial biopsy has been clinically performed, where specimens are obtained via a catheter with a biopsy forceps for pathological and viral testing. This can reveal evidence of myocardial inflammation, though there are still unresolved issues regarding the diagnostic criteria for pathological histology and the elimination of artifacts.
bubble_chart Treatment Measures
Since the cause of the disease is unknown, prevention is difficult. Paying attention to cardiac changes and initiating early treatment during viral infections is of practical significance.
Treatment primarily targets clinical manifestations.
1. Rest and avoidance of exertion must be strongly emphasized, especially for those with cardiac enlargement or reduced cardiac function. Long-term rest is advisable to prevent worsening of the condition.
2. The treatment principles for heart failure are the same as for general heart failure, involving the use of cardiotonic drugs, diuretics, and vasodilators. Due to extensive myocardial damage, Rehmannia-based drugs and diuretics are beneficial. In cases of low glomerular filtration, hydrochlorothiazide may be ineffective, and loop diuretics such as furosemide may be required. Vasodilators like angiotensin-converting enzyme inhibitors are also useful, but they should be started at a low dose to avoid hypotension.
In recent years, it has been found that β-blockers are effective for heart failure in this disease. The mechanism may involve the removal of harmful effects from excessive adrenergic nerve excitation in chronic heart failure, where β-receptor density is downregulated to a greater extent in this disease than after myocardial infarction. After β-blocker treatment, the harmful effects of excessive adrenergic nerve excitation are eliminated, and myocardial β-receptor density is upregulated. It is known that β 1 blockers are beneficial. Treatment should begin with a very small dose and then be gradually increased. This approach can prolong patient survival.
3. For arrhythmias, especially symptomatic ones, antiarrhythmic drugs or electrical methods should be used. Aggressive treatment is necessary for rapid ventricular rhythms or high-grade atrioventricular block, which carry a risk of sudden death.
4. Oral anticoagulants or antiplatelet agents can be used to prevent thromboembolic complications.
5. Drugs that improve myocardial metabolism, such as vitamin C, adenosine triphosphate, coenzyme A, cyclic adenosine monophosphate, and coenzyme Q 10 , can be used as adjunctive therapy.
6. For patients with long-term heart failure unresponsive to medical treatment, heart transplantation should be considered. Postoperative care should focus on controlling infections, improving immunosuppression, and managing rejection, with a one-year survival rate exceeding 85%.
The course of this type varies greatly, with the shortest being death within one year of onset and the longest surviving over 20 years. Patients with significant cardiac enlargement, persistent heart failure, or refractory arrhythmias generally have a poor prognosis. Sudden death is not uncommon among many patients.


