| disease | Drug-induced Liver Disease |
| alias | Medicinal Liver, Drug Induced Liver Disease |
Drug-induced liver disease, abbreviated as drug liver, refers to liver damage caused by drugs and/or their metabolites. It can occur in healthy individuals with no previous history of liver disease or in patients with pre-existing severe illnesses, all of which are referred to as drug liver when varying degrees of liver damage result from the use of certain medications. Currently, there are at least 600 drugs known to cause drug liver, with manifestations identical to various human liver diseases. These can present as hepatocyte necrosis, cholestasis, intracellular microvesicular steatosis, chronic hepatitis, cirrhosis, and more.
bubble_chart Pathogenesis
Drugs are metabolized in the liver through a series of drug-metabolizing enzymes (referred to as drug enzymes, including cytochrome P-450, monooxygenase, cytochrome C reductase, etc.) located on the microsomes of the smooth endoplasmic reticulum in hepatocytes, along with coenzyme II (reduced NADPH) in the cytoplasm. These enzymes facilitate oxidation, reduction, or hydrolysis to form corresponding intermediate metabolites (Phase I antagonism). These intermediates then combine with glucuronic acid or other amino acids (Phase II antagonism, i.e., the biotransformation of drugs) to form water-soluble final products, which are excreted from the body. Final metabolites with a molecular weight greater than 200 are excreted through the biliary system into the intestines, while the rest are eliminated via the kidneys.
The mechanisms by which drugs cause liver injury may include: ① The direct toxic effects of drugs and their intermediate metabolites on the liver, which are predictable; ② Allergic reactions of the body to the drugs or idiosyncratic reactions to intermediate metabolites (idiosyncracy), which are immune responses to the drugs, their metabolites, or covalent complexes formed between the drugs/metabolites and liver macromolecules. These types of drug-induced liver injuries are unpredictable.
The disease mechanism of drug-induced liver injury can directly cause liver damage by altering the physical (viscosity) and chemical (cholesterol/phospholipid ratio) properties of hepatocyte membranes, inhibiting K+, Na+-ATPase on the cell membrane, interfering with the uptake process of hepatocytes, disrupting cytoskeletal functions, or forming insoluble complexes in the bile. It can also selectively damage cellular components, covalently bind to key molecules, or interfere with specific metabolic pathways or structural processes, indirectly leading to liver injury.bubble_chart Pathological Changes
The pathological classification is shown in Table 1.
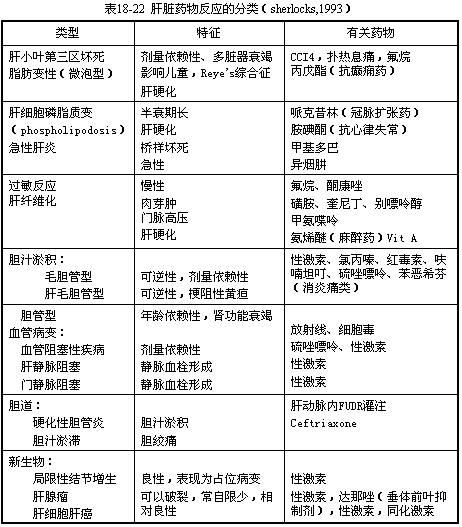
(1) Hepatocyte degeneration and necrosis Hepatocyte injury is the primary manifestation of drug-induced liver injury (DILI). It is mainly caused by toxic intermediate metabolites. The chemical reactions of drug-metabolizing enzymes activate structurally stable drugs, forming electrophilic intermediate metabolites. These highly reactive alkylating, arylating, and acylating substances covalently bind to vital macromolecules (DNA, RNA) within hepatocytes, leading to hepatocyte necrosis. Another pathway involves free radicals generated by P450 oxidation reactions, which covalently bind to proteins or unsaturated fatty acids on the cell membrane, producing lipid peroxides that cause cell membrane injury. Both pathways disrupt Ca2+ homeostasis, leading to membrane pump failure, mitochondrial dysfunction, cytoskeletal damage, and ultimately cell death. Necrosis predominantly occurs in zone 3 of the hepatic lobule (centrilobular region) because this area has the highest concentration of drug-metabolizing enzymes and the lowest oxygen content in the hepatic sinusoids. Drugs such as carbon tetrachloride, acetaminophen, and halothane primarily cause zone 3 (centrilobular) necrosis, accompanied by scattered fatty degeneration but minimal inflammatory response. Other drugs, such as aspirin, NSAIDs, thiazide diuretics, niacin, clofibrate, gemfibrozil (a lipid-lowering agent), oxacillin, sulfonamides, rifampin, ketoconazole, 5-FU, zidovudine (an antiviral), isoniazid, and methyldopa, can cause diffuse parenchymal injury resembling viral hepatitis. This includes hepatocyte damage ranging from spotty necrosis to periportal or bridging necrosis or multilobular necrosis, along with mononuclear cell infiltration in the portal and periportal areas. In contrast, the antiepileptic drug valproic acid and intravenous tetracycline can induce extensive microvesicular steatosis in hepatocytes and liver failure, similar to the findings in Reye syndrome and pregnancy-related fatty liver disease.
(II) Intrahepatic cholestasis is the result of the destruction of the liver cell's gall fel secretion function by drugs and their metabolites, preventing the excretion of gall fel from the cells (intralobular cholestasis), or due to the slowing of gall fel flow in the bile ducts and the progressive destruction and reduction of interlobular bile ducts caused by immune reactions, leading to the accumulation of gall fel in the liver (interlobular cholestasis). Pathologically, it can be divided into three types. ① Canalicular type: Also known as simple cholestasis. Under light microscopy, the liver lobule structure is normal, with only central lobular cholestasis (bile pigment deposition in hepatocytes, bile thrombi in the bile canaliculi). The mechanism of intralobular cholestasis involves the sustained reduction of bile salt-independent gall fel flow by inhibiting Na+, K+-ATPase activity (e.g., estrogen-like substances), decreased lipid fluidity of the hepatic sinusoidal membrane (e.g., S-adenosylmethionine), increased permeability of tight junctions in the intercellular matrix, leading to gall fel water leakage, thickening of gall fel, destruction of the hepatocyte cytoskeleton, and failure of microfilament contraction around the bile canaliculi. Drugs causing such changes mainly include C-17 alkylated testosterone, as well as n-19-nor-17α-ethyltestosterone, methyltestosterone, nandrolone, megestrol, oxymetholone, danazol, etc. Cyclosporine A also induces this type of cholestasis by inhibiting microvesicle transport. Oral contraceptives rarely cause cholestasis nowadays due to reduced hormone content. ② Hepato-canalicular type: Predominantly cholestasis accompanied by grade I hepatocyte damage. Under light microscopy, gall fel stasis is observed in the bile canaliculi, hepatocytes, and Kupffer cells, mainly in the central lobule, with significant cellular reactions (minor mononuclear cell infiltration, predominantly eosinophils). Hepatocytes exhibit feathery degeneration, ballooning degeneration, and focal necrosis. Grade I often involves immune-mediated liver injury, overlapping allergic reactions and drug toxicity. The representative drug is chlorpromazine, a cationic detergent that, due to enterohepatic circulation, reaches high concentrations in gall fel along with its metabolites. It reduces membrane fluidity, inhibits Na+-K+-ATPase, decreases bile flow, inserts into the cell membrane, alters the bilayer lipid structure of the cell membrane, and disrupts the cytoskeleton, leading to bile canaliculi dilation and diverticulum formation. The direct hepatotoxicity of chlorpromazine is related to the generation of free radicals. Other drugs causing cholestasis include phenothiazines, tricyclic antidepressants, erythromycin, carbamazepine, cyproheptadine, tolbutamide (D860), captopril, phenytoin sodium, SMZ-TMP, sulfasalazine (SASP), imipramine, benzimidazole, organic arsenic, etc. The above two types anatomically belong to intralobular cholestasis. ③ Bile duct type: Also known as interlobular cholestasis, where the bile ducts and canaliculi are filled with highly concentrated bile thrombi (complexes of bilirubin and drug metabolites) without surrounding inflammation. Benoxaprofen belongs to this category but has been banned due to its association with renal failure.(3) Mixed Type The pathology is mainly characterized by hepatic parenchymal damage accompanied by grade I cholestasis, and may also involve extrahepatic manifestations such as fever, rash, lymphadenopathy, myocarditis, and interstitial nephritis. These changes are mostly due to the body's allergic reaction to drugs, triggered by immune mechanisms. Common drugs include phenytoin sodium, quinidine, allopurinol, and nitrofurantoin. In allergic liver damage, drugs acquire antigenicity in the form of hapten complexes, sensitizing T cells and producing cytotoxic T cells and antibody-dependent cell-mediated cytotoxicity (ADCC). Alternatively, metabolites with electrophilic groups or free radicals may bind to liver cell proteins, forming new antigens and inducing an immune response.
(4) Chronic Hepatitis Drugs confirmed to cause chronic hepatitis include diacetylphenol (continued use after liver damage may progress to cirrhosis), methyldopa, nitrofurantoin, dantrolene (a skeletal muscle relaxant), isoniazid, propylthiouracil, sulfonamides, and halothane. Histological changes resemble those of autoimmune chronic hepatitis or chronic sexually transmitted disease-related hepatitis, including periportal mononuclear cell infiltration, accompanied by six pulse conditions such as periportal, bridging, and multilobular necrosis.
Additionally, pathological drug-induced liver injury may include the following rare types of liver damage: ① Vascular lesions: hepatic sinusoidal dilatation and peliosis hepatis, hepatic vein and portal vein obstruction (sex hormones); ② Sclerosing cholangitis (intrahepatic stirred pulse perfusion of cytotoxic drugs such as 5-fluorodeoxyuridine FUDR); ③ Induction of liver tumors (sex hormones, danazol).
bubble_chart Clinical Manifestations
One of the clinical manifestations of drug-induced liver injury is related to the type of hepatotoxic drugs and the mechanism of liver disease. Based on clinical features, it can be divided into acute and chronic categories. Among acute hepatocellular injuries, acute drug-induced hepatitis is the most common. When hepatocellular necrosis predominates, the clinical presentation closely resembles acute viral hepatitis, often featuring fever, fatigue, poor appetite, jaundice, and elevated serum transaminase levels (2–30 times the normal range). ALP and albumin are less affected, while hyperbilirubinemia and prolonged prothrombin time are associated with severe liver damage (Grade III). In milder cases, discontinuation of the drug leads to recovery within weeks to months. In severe cases, liver failure may occur, presenting with progressive jaundice, bleeding tendencies, and hepatic encephalopathy, often resulting in death.
Acute drug-induced liver injury primarily caused by allergic reactions often presents with fever, rash, jaundice, and lymphadenopathy, accompanied by Grade II elevations in serum transaminase, bilirubin, and ALP levels. The history of drug exposure is usually short (within 4 weeks).
Drug-induced liver injury dominated by cholestasis exhibits clinical and laboratory findings similar to intrahepatic cholestasis, extrahepatic biliary obstruction, or acute cholangitis, including fever, jaundice, upper abdominal pain, pruritus, right upper quadrant tenderness, and hepatomegaly. Serum transaminase levels are mildly elevated, while ALP is significantly increased (2–10 times), along with markedly elevated conjugated bilirubin (34–500 μmol/L), bile salts, lipoprotein X, GGT, and cholesterol. Anti-mitochondrial antibodies are negative. Recovery typically occurs within 3 months to 3 years after drug discontinuation, though a minority may develop vanishing bile duct syndrome with a chronic progressive course. Occasionally, bile duct damage is irreversible, progressing to cirrhosis.
Drug-induced chronic hepatitis shares clinical features with autoimmune chronic hepatitis, ranging from mild or asymptomatic to severe cases with liver failure and hepatic encephalopathy. Its generation and transformation resemble chronic viral hepatitis, with elevated serum transaminase and G-GT levels. Progressive cases may lead to cirrhosis accompanied by hypoalbuminemia and coagulation disorders.
The diagnosis of drug-induced liver injury (DILI) can be made comprehensively based on medication history, clinical symptoms, blood tests, liver function tests, liver biopsy, and the effects of drug withdrawal. Before diagnosing DILI, the following should be assessed: ① Medication history: For any patient with liver disease, it is necessary to inquire about medications taken within the three months prior to the onset of symptoms, including dose, route of administration, duration of use, and concurrent use of other drugs. ② Whether there was pre-existing liver disease, evidence of viral hepatitis, or other causes of liver disease; ③ Whether the primary disease could potentially involve the liver; ④ Any history of drug allergies or allergic diseases. In addition to medication history, identifying any related allergic reactions, such as rashes and eosinophilia, is crucial for diagnosing DILI. When diagnosing DILI, the following conditions should be differentiated: viral hepatitis, systemic bacterial infections, postoperative intrahepatic cholestasis, cholangitis with or without acute pancreatitis, bile duct injury, congestive heart failure, and worsening liver function in chronic liver disease.
The diagnostic criteria for liver disease caused by drug hypersensitivity are: ① Onset of liver dysfunction within 1–4 weeks after starting the medication; ② Initial symptoms primarily include fever, rash, pruritus, and jaundice; ③ In the early stage of the disease, peripheral blood eosinophilia (≥6%) or leukocytosis is observed; ④ Positive drug sensitivity tests (lymphocyte culture test, skin test); ⑤ Re-exposure to the drug may occasionally re-trigger liver disease. A definitive diagnosis can be made if criteria ① and ④ or ① and ⑤ are met; a presumptive diagnosis can be made if criteria ① and ② or ① and ③ are met. Liver biopsy in the early stages of the disease can help differentiate the type of lesion and assess the extent of damage.
bubble_chart Treatment Measures
Discontinuing drugs that cause drug-induced liver injury or potentially induce it is the most crucial treatment. Adequate rest, enhanced nutrition, and supportive therapy should be provided, along with a high-protein, high-carbohydrate, low-fat diet, supplemented with vitamins C, B, and E. The use of reduced glutathione to replenish SH groups in the liver facilitates the biotransformation of drugs.
For cholestasis, phenobarbital may be tried to promote the production of transport proteins Y and X in hepatocytes, converting indirect bilirubin into direct bilirubin and improving bilirubin metabolism. Patients with severe cholestasis can take cholestyramine 30mg twice daily (morning and evening) to reduce the reabsorption of bile acids and drugs in the gastrointestinal tract. Short-term use of glucocorticoids and Stronger Neo-Minophagen C (80–100ml added to glucose solution for intravenous drip, once daily) may also be considered. Quantum blood therapy, administered twice weekly for 1–2 months, may have a bilirubin-lowering effect in some cases of refractory cholestasis.
Recent studies suggest that 5-adenosyl-L-methionine, also known as adenosylmethionine (Ademetionine, SAMe), is an effective drug for treating intrahepatic cholestasis. Animal experiments have shown that this drug can prevent and reverse cholestasis induced by ethinyl estradiol, alpha-naphthyl isothiocyanate, and chlorpromazine. Clinical trials (double-blind, multicenter, and meta-analysis) have confirmed that Yaodui SAMe has significant efficacy in alleviating itching, improving quality of life, and normalizing generation and transformation indicators in intrahepatic cholestasis caused by various factors, with no side effects. Dosage: 1–2g/day intravenously for 2 weeks, followed by 1.6g/day orally in two divided doses until symptoms and generation and transformation indicators improve, typically lasting 4–8 weeks.
SAMe is a compound synthesized from methionine and ATP under the action of SAMe synthase, playing a critical role in transmethylation and transsulfuration. Through transmethylation, it increases the biosynthesis of membrane phospholipids. As the phospholipid/cholesterol ratio rises, membrane fluidity improves, and K+, Na+-ATPase activity is enhanced, promoting bile acid transport. Simultaneously, through transsulfuration, it boosts the production of glutathione and cysteine, the primary intracellular detoxifying agents, enhancing hepatocyte detoxification and protection against free radicals. The resulting taurine can bind with bile acids, increasing their solubility. Thus, SAMe has a certain preventive and therapeutic effect on intrahepatic cholestasis. In liver injury, SAMe synthase activity declines, reducing endogenous SAMe production. Therefore, supplementing with SAMe as a drug can address this deficiency and provide therapeutic benefits.
For liver injury caused by excessive paracetamol, N-acetylcysteine (N-AC) can be administered. This precursor of cysteine should be given at 140mg/kg (orally or via gastric tube) after gastric lavage within 24 hours of drug ingestion, followed by 70mg/kg every 4 hours for a total of 72 hours.
In severe cases leading to liver failure or grade III cholestasis progressing to cirrhosis, liver transplantation should be considered.
Most patients can recover after stopping the medication, showing clinical and histological improvement, with the fastest recovery taking only a few weeks and the slowest requiring several years. A small number of patients may experience severe and extensive liver injury, leading to fulminant liver failure or progression to cirrhosis, which can be fatal without a liver transplant.


