| disease | Congenital White Internal Visual Obstruction |
| alias | Infant White Internal Visual Obstruction |
Congenital cataract is a common eye disease in children. The partial or complete opacification of the lens occurring within the first year after birth is called congenital cataract. Since factors causing lens opacification are already present at birth but cataracts have not yet developed, and the lens opacity occurs within the first year of life, congenital cataract is also known as infantile cataract. Congenital cataracts can be familial or sporadic; they may affect one or both eyes; and they may be associated with other ocular abnormalities. Additionally, various genetic or systemic diseases can also be accompanied by congenital cataracts. However, the most common presentation is cataracts as the sole abnormality. This condition has many types with different etiologies. To confirm the diagnosis, necessary laboratory tests may sometimes be required. Because congenital cataracts can lead to deprivation amblyopia at an early stage, their treatment differs from that of typical adult cataracts.
bubble_chart Epidemiology
Congenital white internal visual obstruction is a relatively common childhood eye disease, but there has been no statistical data on its prevalence in our country in the past. In recent years, through surveys of blinding eye diseases and hereditary eye diseases, it has been concluded that the group prevalence of congenital white internal visual obstruction in China is 0.05% (1:1918), which is lower than the prevalence abroad (Francois, 0.4%).
Since this disease is an important cause of blindness and amblyopia in children, efforts should be made to reduce the prevalence of congenital white internal visual obstruction from the perspectives of eugenics, better child-rearing, and blindness prevention. Investigations into the causes of blindness among blind children in Tianjin, Shanghai, and Beijing found that 22–30% of blind children were blinded due to congenital white internal visual obstruction, ranking as the second leading cause of blindness. Additionally, many children suffer from irreversible amblyopia due to this condition. Francois reviewed previous data and found that congenital white internal visual obstruction accounts for 10.0%–38.0% of the causes of childhood blindness.
bubble_chart EtiologyWhen a newborn has white internal visual obstruction in one or both eyes, the disease cause should be clearly identified. If there is a positive family history, it is related to heredity. Additionally, environmental factors also contribute to its onset, and some cases are complications of systemic diseases. Even after pedigree analysis or laboratory tests, the disease cause cannot be identified in one-third of cases. Such cases of white internal visual obstruction with unknown causes are referred to as idiopathic white internal visual obstruction. Patients have no other ocular abnormalities or systemic diseases, and the lens opacity may be caused by multiple factors. It is speculated that one-fourth of this group of diseases is caused by new autosomal dominant gene mutations. Some are related to systemic diseases, but due to the mild manifestations of these systemic diseases, they are overlooked or unrecognized.
1. Heredity Over the past 50 years, there has been more in-depth research on the heredity of congenital white internal visual obstruction. Approximately one-third of congenital white internal visual obstruction cases are hereditary (Figures 2 and 3). Among these, autosomal dominant inheritance is the most common. Statistical data from our country indicate that dominant inheritance accounts for 73%, recessive inheritance for 23%, and no reports of sex-linked inheritance have been found. In regions or countries with high rates of consanguineous marriages, recessive inheritance is not uncommon.
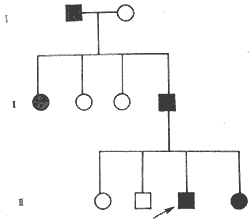
Figure 2: Autosomal dominant inheritance pedigree (nuclear white internal visual obstruction)
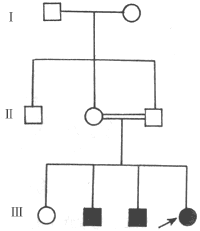
Figure 3: Autosomal recessive inheritance pedigree (complete white internal visual obstruction)
Autosomal recessive hereditary white internal visual obstruction is relatively rare and is often associated with consanguineous marriages. The incidence rate in offspring of consanguineous marriages is more than 10 times higher than that in offspring of random marriages. The most common type is nuclear white internal visual obstruction. Misclassification can also occur in recessive hereditary white internal visual obstruction because, in families with random marriages, if the parents have a normal phenotype but are carriers of the white internal visual obstruction pathogenic gene, and one of their children has congenital white internal visual obstruction, it may be mistakenly attributed to white internal visual obstruction of unknown cause.
Since there is currently no method to detect recessive gene carriers, prohibiting consanguineous marriages is an important measure to reduce recessive hereditary white internal visual obstruction. It is now known that this type of white internal visual obstruction involves 2–3 pathogenic genes.

X-linked recessive hereditary white internal visual obstruction is even rarer, with one pathogenic gene. Affected males mostly have nuclear white internal visual obstruction, which remains stationary or gradually progresses to mature white internal visual obstruction. Female carriers exhibit Y-suture opacities without visual impairment.
Hereditary congenital white internal visual obstruction has been observed in animal experiments, such as autosomal dominant nuclear white internal visual obstruction in guinea pigs and recessive hereditary Y-suture white internal visual obstruction in rabbits, both of which can gradually progress to complete white internal visual obstruction.
Sporadic white internal visual obstruction without a family history may be caused by a mutation in an autosomal dominant gene. The patient is the first-generation white internal visual obstruction case in this family, and their children will have a 50% chance of inheriting the mechanism of disease.
In molecular genetic studies of congenital cataract, the findings have been inconsistent due to the involvement of different gene loci and polymorphisms. Research on DNA recombination technology has revealed that defects in the γ gene of the lens can cause hereditary congenital cataract. Other scholars suggest that cataract formation is associated with defects in the crystallin gene on chromosome 21 and the MIP protein gene on chromosome 12. Studies using laser Raman spectroscopy on hereditary cataract have shown that during the initial formation of cataract, changes occur in tyrosine residues of the lens, with sulfhydryl groups converting into disulfide bonds, forming cross-linked macromolecular polymers that stabilize the opaque lens protein aggregates.
2. Non-hereditary Apart from heredity, environmental factors are another major cause of congenital cataract, accounting for about one-third of cases.
It should be noted that maternal infections during the first two months of pregnancy are a significant factor contributing to cataract formation. Infections during pregnancy (such as rubella, chickenpox, herpes simplex, measles, herpes zoster, and influenza viruses) can lead to fetal lens opacities. At this stage, the lens capsule is not fully developed and cannot defend against viral invasion. Additionally, the lens protein synthesis is highly active, making it sensitive to viral infections, which disrupts the growth and development of lens epithelial cells. Concurrent nutritional and biochemical changes, along with metabolic disturbances in the lens, result in opacities. By the late stage of pregnancy (third trimester), the fetal lens capsule has matured sufficiently to protect the lens from viral damage.
Among the various viral infections causing cataract, rubella virus infection is the most common. During the 1964–1965 rubella epidemic in the United States, 20,000 children developed rubella syndrome, 50% of whom had congenital cataracts at birth or within the first year of life. The severity of opacities correlates with the timing and extent of viral invasion of the lens. Moreover, with the rising incidence of sexually transmitted diseases, herpes simplex virus type II (HSV-2)-induced cataracts warrant attention. Newborns can contract the virus from the maternal birth canal. HSV-2 has been cultured from the lens cortex of affected patients. While the lens may initially appear clear, cataracts develop rapidly thereafter.
Maternal malnutrition during pregnancy, pelvic radiation exposure, certain medications (e.g., high-dose tetracycline, hormones, salicylates, anticoagulants), systemic diseases during pregnancy (heart disease, nephritis, diabetes, anemia, hyperthyroidism, tetany, calcium metabolism disorders), and vitamin D deficiency can all contribute to fetal lens opacities.
Another common cause of congenital cataract is developmental disturbances in the last three months of fetal life. Typical manifestations include low birth weight, hypoxia, and central nervous system damage in premature infants. Animal studies have confirmed that intrauterine hypoxia can lead to congenital cataract.
Approximately 2.7% of premature infants develop cataracts after birth, presenting with clear vesicles under the anterior and posterior lens capsules, bilaterally symmetrical. These vesicles may resolve spontaneously or progress to diffuse posterior subcapsular opacities. Additionally, immature premature infants often require high-concentration oxygen therapy, which increases the risk of retinopathy of prematurity (ROP), followed by lens opacities months later.
In summary, environmental factors play a crucial role in non-hereditary congenital cataracts. Therefore, emphasizing perinatal care is essential to reduce the incidence of congenital cataracts.
3. Sporadic About one-third of congenital cataracts are of unknown cause, classified as sporadic, with no clear environmental influence. Some cases in this group may still be hereditary, such as new autosomal dominant mutations presenting as cataracts in the first generation without a family history, making it difficult to confirm heredity. Isolated cases of recessive inheritance are also challenging to diagnose as hereditary.
bubble_chart Clinical Manifestations
Congenital cataracts have many types, which can be complete or incomplete, and can also be classified as nuclear, cortical, or membranous cataracts. Depending on the location, morphology, and severity of the opacity, the degree of visual impairment varies. The more common types of cataracts include the following.
1. **Total cataract** The lens is entirely or nearly entirely opaque, or it may gradually develop after birth, becoming fully opaque within the first year of life. This occurs due to damage to the lens fibers during the intermediate stage (second stage) or late stage (third stage) of development. Clinically, the lens in the pupillary area appears white and opaque, sometimes with thickened or calcified capsules, or even cortical condensation and dislocation. Visual impairment is significant, and the condition is often bilateral. Autosomal dominant inheritance is most common, with the trait passing through multiple generations in a family (see Figure 3). A minority of cases are recessive, and very few are X-linked recessive.
2. **Membranous cataract** When the lens fibers of a congenital total cataract undergo degenerative changes in utero, the cortex is gradually absorbed, forming a membranous cataract. Swelling of the cortex or traction from vitreous pulsation on the posterior capsule may cause rupture of the posterior capsule, accelerating cortical absorption and resulting in congenital aphakia. Clinically, it presents as a gray-white, hard membrane with irregular surfaces and variably colored spots. Sometimes, ciliary processes and blood vessels can be seen on the membrane surface, possibly originating from embryonic vascular membranes. Fibrous tissue may also extend onto the membrane surface, hence it is also called vascular membranous cataract or fibrous cataract. It can occur unilaterally or bilaterally, with severe visual impairment. A few cases are associated with intrauterine iridocyclitis.
3. **Nuclear cataract** This type is relatively common, accounting for about one-fourth of congenital cataracts. Both the embryonic and fetal nuclei are affected, presenting as dense white opacities (see Color Figure 4), covering an area of 4–5 mm and completely obscuring the pupillary zone, leading to significant visual impairment. It is usually bilateral, with autosomal dominant inheritance being most common, though recessive inheritance and sporadic cases also occur.
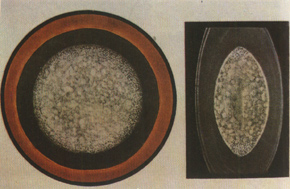
**Figure 4: Nuclear cataract**
4. **Central pulverulent cataract** This results from damage to the embryonic nucleus during the first three months of gestation, while the fetal nucleus remains unaffected. Clinically, there is a dust-like or granular opacity between the two Y-sutures of the embryonic nucleus, hence it is also called lamellar pulverulent cataract. If the fetal nucleus is also affected, it manifests as a nuclear or lamellar cataract. Under slit-lamp examination, numerous fine white dots are visible within the opacity, covering an area of about 1–2.5 mm. It is usually bilateral, stable, and has minimal impact on vision.
5. **Perinuclear cataract** This is a very common type, accounting for 40% of congenital cataracts. Since the opacity lies between the layers surrounding the nucleus, it is also called lamellar cataract. It is typically non-progressive and bilateral. Clinically, the opacity encircles the fetal nucleus, composed of many fine white dots, while the cortex and embryonic nucleus remain clear. On the periphery of the opacity, "V"-shaped opacities straddle the anterior and posterior parts of the opacity band, known as "riders." Since the central nucleus remains clear (see Color Figure 5), visual impairment is not severe. This condition arises from metabolic disturbances in the lens during a specific embryonic period, leading to a layer of opacity. It may also be associated with systemic diseases. Autosomal dominant inheritance is most common, with reports of vertical transmission in one family spanning up to 11 generations, with 132 out of 542 individuals affected by perinuclear cataract.
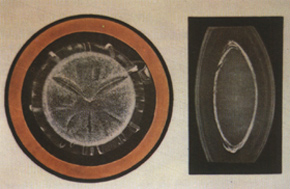
Figure 5 Nuclear cataract internal visual obstruction
6. Anterior axial embryonic cataract internal visual obstruction (anterior axil embryonic cataract) This type of cataract internal visual obstruction is also a relatively common congenital cataract internal visual obstruction, accounting for about 25%. Behind the anterior Y-suture, there are numerous chalky fragment-like or white crystalline opacities. These opacities form during the first four months of embryonic development. Due to their localized nature, they do not significantly affect vision, and thus generally do not require treatment.
7. Anterior polar cataract internal visual obstruction (anterior polar cataract) The characteristic of this condition is a localized opacity in the central anterior capsule membrane of the lens. The extent of the opacity varies—it can be as small as a white dot no larger than 0.1 mm or large enough to cover the entire pupillary area. It is often round and may extend into the lens cortex or protrude into the anterior chamber. In some cases, the protruding anterior polar portion may even touch the cornea, termed a pyramidal cataract (pyramidal cataract). A corresponding white localized opacity is present in the central cornea. Some cases may also exhibit iris remnants (Figure 6). The lens nucleus in anterior polar cataract internal visual obstruction remains transparent, indicating damage to the capsule membrane during the late embryonic stage [third stage]. An abnormal reaction of the capsule membrane forms a white mass, which can be removed with a needle while preserving the integrity of the lens capsule membrane. The condition is bilateral, non-progressive, and does not significantly affect vision, so treatment is usually unnecessary.
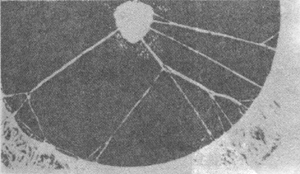
Figure 6 Anterior polar cataract internal visual obstruction and iris remnants
8. Posterior polar cataract internal visual obstruction (posterior polar cataract) The hallmark of this condition is a localized opacity in the central posterior capsule membrane of the lens, with irregular edges and varying shapes—disk-like, nuclear, or calyx-like. It is often accompanied by persistent vitreous artery remnants, with the central opacity marking the termination site of the vitreous artery. A minority of cases are progressive, but most remain static. Severe vision impairment is rare. In adolescents, the posterior polar opacity may extend into the cortical region, forming radial opacities that can affect vision to some degree.
9. Sutural cataract internal visual obstruction (sutural cataract) The clinical manifestation of this condition is abnormal calcium deposition along the fetal nuclear Y-suture, appearing as three radial white lines, hence also called tri-radiate cataract (tri-radiate cataract). The Y-suture cataract internal visual obstruction consists of linear, nodular, or branching opacities, appearing greenish-white or blue with irregular edges. It is typically localized and non-progressive, with little impact on vision, so treatment is generally unnecessary. A family history is common, with reports of continuous generational inheritance, suggesting an autosomal dominant pattern. It may coexist with coronary cataract or cerulean cataract.
10. Coralliform cataract internal visual obstruction (coralliform cataract) Coralliform cataract is relatively rare. A round or rectangular gray or white opacity appears in the central lens region, radiating outward to the capsule membrane, resembling a cluster of forward-growing coral (Figure 7). The central nucleus is also opaque, which may affect vision to some extent. The condition is usually static and non-progressive, often with a family history, following an autosomal dominant or recessive inheritance pattern.

Figure 7 Coralliform cataract internal visual obstruction
11. Punctate cataract internal visual obstruction (punctate cataract) White, bluish-green, or light brown punctate opacities appear in the lens cortex or nucleus, occurring after birth or during adolescence. The opacities are static and non-progressive, generally not affecting vision or causing only grade I vision impairment. Sometimes, they may coexist with other types of opacities (Figure 8).
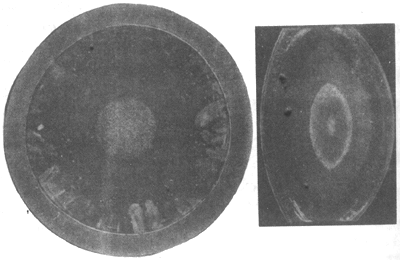
Figure 8 Central punctate cataract with coronary cataract (frontal and lateral views)
12. Disciform cataract (disciform cataract) This condition was discovered by Nettleship and others in the Coppock family, where several members had congenital cataracts, hence it is also known as Coppock cataract. Its characteristic feature is a clearly defined disc-shaped opacity between the nucleus and the posterior pole, with clear cortex separating the opaque area from the posterior pole. Due to the small extent of opacity, it does not affect vision. The lens opacity occurs in the fourth month of embryonic development and may be related to localized metabolic abnormalities in the lens.
13. Disc-shaped cataract (disc-shaped cataract) Disc-shaped cataract is relatively rare. The pupillary area of the lens exhibits dense opacity, with central calcification and thinning, presenting as a flat disc shape, hence the name disc-shaped cataract (Color Figure 9). Since the lens lacks a nucleus, the central part becomes even thinner, resembling a dumbbell shape in cross-section. There is a clear hereditary tendency.
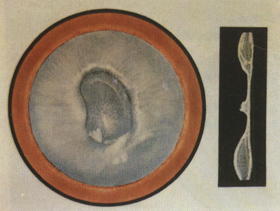
Figure 9 Disc-shaped cataract
14. Hard nucleus liquefaction cataract Hard nucleus liquefaction cataract is very rare. Due to liquefaction of the peripheral lens fiber layers, there is a translucent milky fluid within the lens capsule, with the brown embryonic nucleus floating in the liquefied cortex. Sometimes the nucleus also liquefies. When the cortex liquefies, the capsule may be damaged, reducing permeability. The release of lens proteins can irritate the ciliary body, or the floating nucleus may stimulate the ciliary body, potentially leading to uveitis or glaucoma.
bubble_chart Auxiliary Examination
After birth, a white reflection in the pupil area of a newborn is called leukocoria (1eukocoria), among which the most common is congenital white internal visual obstruction, and other eye diseases can also cause it. Due to differences in clinical manifestations, treatment, and prognosis, timely and accurate differential diagnosis is very necessary.
1. Premature labor retinal membrane lesions (retrolental fibroplasia). This disease occurs in low-birth-weight premature infants, and inhalation of high concentrations of oxygen may be the cause. Both eyes are affected. The retinal membrane vessels are dilated and tortuous, with neovascularization and edema in the peripheral retina. Fibrovascular tissue behind the lens pulls the ciliary body toward the center, resulting in white internal visual obstruction and retinal detachment.
2. Persistent hyperplastic primary vitreous. The affected child is usually full-term and delivered normally, with the condition mostly unilateral. The affected eye is small, the anterior chamber is shallow, the lens is relatively small, and the ciliary processes are very long, reaching the posterior pole of the lens. Behind the lens, there is a vascular fibrous membrane with abundant blood vessels. The posterior pole of the lens is cloudy, and the iris-lens diaphragm is displaced forward.
3. Inflammatory pseudoglioma. Most cases are bilateral, with a few being unilateral. There is a white patch behind the lens, the eyeball becomes smaller, and intraocular pressure decreases. The cause is maternal infection during the last three months of embryonic development in the uterus or neonatal endophthalmitis after birth.
4. Retinal membrane blastoma. This is the most common intraocular malignant tumor in children. Although it mostly occurs before the age of 2–3, it can also appear very early, with white pupils visible within days after birth. The tumor is milky white or yellowish-white, and when it grows to a certain size, light entering the eye reflects as yellowish-white. Continued tumor growth causes retinal detachment, calcification spots on the surface, elevated intraocular pressure, and eventually secondary glaucoma and extraocular metastasis.
5. External exudative retinal membraneitis (Coats disease). The retinal membrane has white-yellow lesions, grade I elevation, with neovascularization and microaneurysms on the surface, and capillary dilation. In severe cases, extensive retinal detachment results in white pupillary reflection. Advanced stages show iris neovascularization, secondary glaucoma, and iridocyclitis.
6. Retinal membrane dysplasia. The affected child is usually full-term and delivered normally, with small eyeballs, a very shallow anterior chamber, and a white tissue mass behind the lens causing white pupils. It is often associated with cerebral dysplasia, congenital heart disease, cleft palate, and polydactyly.
7. Congenital toxoplasmosis. This disease has been reported in China in recent years. Its characteristic is recurrent intraocular inflammation, eventually leaving pigmented scars in the choroidal membrane and retina, mostly in the macular area, resulting in white pupillary reflection. It may also present with hepatosplenomegaly, jaundice, hydrocephalus, and cerebral calcification. A positive toxoplasma indirect hemagglutination test and indirect immunofluorescence antibody test can confirm the diagnosis.
8. Toxocariasis. The fundus of affected children shows granuloma formation, clinically divided into two types: one is a localized choroidal membrane retinal granuloma in the posterior pole without active inflammation, and the other is vitreous opacity with obvious inflammation. Both can cause white pupillary reflection. A history of contact with animals (cats or dogs) is often reported.
Other rare causes of leukocoria include Norrie disease, posterior pole coloboma, vitreous hemorrhage organization, and severe retinal membrane gliosis.
bubble_chart Treatment Measures
Congenital cataracts have diverse clinical manifestations and varying etiologies. They may affect one or both eyes, present as complete or incomplete lens opacities, and may be accompanied by amblyopia. Consequently, their treatment differs from that of adult cataracts.
1. **Amblyopia** The goal of treating cataracts in children is to restore vision, with the primary focus on preventing deprivation amblyopia. The opaque lens obstructs normal retinal stimulation, disrupting the development of the visual system and leading to deprivation amblyopia. Studies show that covering both eyes of newborn monkeys for 12 weeks results in irreversible amblyopia, with permanent neuronal degeneration from the lateral geniculate nucleus to the cerebral cortex. The timing of amblyopia onset in monkeys aligns with the onset of nystagmus in children with cataracts, suggesting that 12 weeks is the critical period for severe, irreversible amblyopia. Existing data indicate that treating deprivation amblyopia before 4 months of age is reversible, whereas treatment after 6 months yields poor outcomes.
Deprivation amblyopia may be unilateral or bilateral. If amblyopia develops, infants as young as 2–3 months may exhibit nystagmus, indicating a failure to establish fixation reflexes. Therefore, early intervention for congenital cataracts is essential to ensure normal development of fixation reflexes.
2. **Conservative Treatment** Bilateral incomplete cataracts with vision above 0.3 may not require surgery. However, since infants cannot undergo visual acuity testing, if the cataract is centrally located but the peripheral fundus remains visible, surgery may be deferred. Long-term use of mydriatics can be considered until vision can be assessed to determine the need for surgery. However, atropine-induced mydriasis causes accommodative paralysis, necessitating corrective glasses for reading.
It is important to note that visual acuity correlates more closely with the density of lens opacity than with its extent. For example, a 5.5 mm opacity may yield the same visual acuity as a 2.0 mm opacity.
Historically, unilateral incomplete cataracts were not considered for surgery. However, with timely postoperative corrective lenses, occlusion therapy for the healthy eye, or contact lens fitting, satisfactory visual outcomes can still be achieved.
3. **Surgery**
(1) **Preoperative Evaluation**
**Ocular Examination** Assessing vision in children under 3–4 years old is challenging. Instead, fixation reflexes and responsiveness to the environment can provide preliminary insights. Regular slit-lamp and fundus examinations are necessary to monitor the progression or regression of lens opacities.
**Systemic Evaluation** Concomitant systemic abnormalities should be ruled out. Consultation with specialists is recommended to exclude cardiovascular or central nervous system disorders, minimizing anesthesia risks.
Additionally, a thorough family and genetic history aids in diagnosis and prognosis assessment.
(2) **Timing of Surgery** The optimal timing for surgery depends on the type of cataract.
**Bilateral Complete Cataracts** Surgery should be performed within 1–2 weeks after birth, with a maximum delay of 6 months. The second eye should be operated on within 48 hours or sooner after the first eye to prevent deprivation amblyopia due to unilateral occlusion.
**Bilateral Incomplete Cataracts** If vision is 0.1 or worse with no fundus visibility, early surgery is recommended. If the peripheral fundus is visible, surgery may be deferred.
Unilateral complete cataract internal visual obstruction. In the past, it was widely believed that vision could not be restored after surgery for unilateral complete cataract internal visual obstruction, as 30–70% of cases are complicated by other ocular abnormalities (microphthalmos, ocular tremor, strabismus, and certain fundus diseases), along with pre-existing amblyopia. However, recent clinical data indicate that if surgery is performed during the neonatal period or even within 7 hours after birth, followed by bilateral eye patching, fitting of contact lenses (26.00–30.00D) on the 4th day, and regular follow-ups until visual acuity can be assessed, many affected eyes can still achieve vision better than 0.2. If surgery is performed after the age of 1, even if the procedure is highly successful and the pupillary area remains clear, it is difficult for vision to reach 0.2. Therefore, it is particularly emphasized that unilateral cataract internal visual obstruction must be treated with early surgery, prompt optical correction, and strict amblyopia prevention measures.
Children with rubella syndrome should not undergo surgery too early. This is because in the early stages after infection, the rubella virus still exists in the lens. During surgery, the latent virus in the lens may be released, causing iridocyclitis, and in 5-20% of cases, eyeball atrophy may occur postoperatively due to inflammation. In rubella syndrome, the white internal visual obstruction is often centrally opaque with clear peripheral cortex, making optical iridectomy a viable option.
(3) Surgical Methods: Since Scheie improved the technique of white internal visual obstruction aspiration in 1960, this procedure has been widely used for treating congenital white internal visual obstruction. This surgery is simple and safe, and can be performed on newborns shortly after birth. Optical iridectomy has certain limitations, while linear extraction and needling procedures are rarely used nowadays.
Optical Iridectomy: Suitable for cases where vision improves after pupil dilation, such as lamellar white internal visual obstruction with small areas of opacity, nuclear white internal visual obstruction, or anterior/posterior polar white internal visual obstruction. After iridectomy, the size and position of the pupil are altered. The incision is typically made in the superotemporal quadrant, as the upper eyelid covers it, minimizing cosmetic impact.
White Internal Visual Obstruction Aspiration: Performed under general anesthesia using an operating microscope. The pupil is fully dilated with 1% atropine, and a corneal incision of about 2mm is made. A cystotome or needle is inserted into the anterior chamber to thoroughly rupture the anterior lens capsule, followed by aspiration of the anterior capsule and cortex using an irrigation-aspiration needle.
Aspiration preserves the integrity of the posterior lens capsule, but epithelial cells quickly grow from the periphery toward the center postoperatively. Within weeks, the posterior capsule becomes translucent, affecting retinal imaging. Therefore, it is now recommended to use a vitrectomy cutter during the same surgery to excise and aspirate the vitreous and posterior lens capsule, a procedure known as lensectomy. In infants and children, the posterior lens capsule is fused with the vitreous, so cutting the posterior capsule also involves cutting the anterior hyaloid membrane. The vitrectomy cutter can be inserted through a corneal incision or a pars plana incision.
4. YAG Laser and Membranous White Internal Visual Obstruction: After congenital white internal visual obstruction aspiration, 90% of cases develop secondary membranes, and over half require surgical incision to improve vision. Since 1982, YAG laser has been widely used for treating membranous white internal visual obstruction where available, offering simplicity, efficacy, and safety. The success rate for a single procedure is 97%, with over 95% of cases showing improved vision post-treatment. YAG laser posterior capsulotomy can be performed one month after aspiration, with a capsular opening diameter of about 3.7mm.
Complications of YAG Laser Treatment: Elevated intraocular pressure, typically occurring 2-4 hours postoperatively and normalizing within 24 hours. Injury to iris vessels or traction on iris-lens capsule adhesions may cause iris hemorrhage or minor anterior chamber bleeding. Capsule fragments entering the anterior chamber or vitreous can lead to grade I pigmentary uveitis. A small percentage (3-9%) may develop cystoid macular edema 6-20 months later. Rare cases may experience retinal holes or detachment. YAG laser can also damage intraocular lenses. Despite these complications, YAG laser remains the best method for treating membranous white internal visual obstruction.
(5) Intraocular Lens Implantation: Choyce (1955) first used anterior chamber intraocular lenses for congenital white internal visual obstruction, but due to numerous
complications, this method is no longer used. Shearin8 (1977) pioneered the use of posterior chamber intraocular lenses, which have since been widely adopted for both adults and children.
The goal of intraocular lens implantation in infants and children is not only to improve vision but also to prevent amblyopia and promote fusion development. However, due to the unique characteristics of pediatric ocular tissues, intraoperative and postoperative complications are significantly higher than in adults. Therefore, this is not a routine procedure and is generally performed no earlier than 2 years of age.
Intraoperative complications Due to the low rigidity of the sclera in infants and young children, there is a tendency for scleral collapse during surgery, especially when the scleral incision is large. In severe cases, there is a risk of intraocular content loss.
Postoperative complications Due to scleral collapse, shallow anterior chamber, and contact between the intraocular lens and corneal endothelium during implantation, linear keratitis may occur. However, since the corneal endothelial activity in infants and children is high, recovery can be achieved within 48 to 72 hours postoperatively. Other complications are the same as those in adults after surgery, such as iridocyclitis, endophthalmitis, bullous keratopathy, cystoid macular edema, glaucoma, etc.
(6) Corneal contact lens: For unilateral congenital cataracts, early surgery and postoperative use of contact lenses are key to preventing amblyopia and restoring vision. If glasses are used for correction after unilateral cataract surgery, the image disparity between the two eyes is 22–35%, whereas with contact lenses, this disparity can be reduced to 8%. Additionally, contact lenses eliminate the prism side effects caused by aphakic glasses (Figure 1), resulting in better peripheral vision and an increased retinal image area. Infants can also wear contact lenses. The drawbacks include difficulties in fitting infants and children, the lenses being prone to loss, deformation, or breakage, and the most significant risk being the potential for suppurative corneal ulcers. Furthermore, due to the small corneal curvature radius in newborns, high plus-power lenses are required, which fit tightly on the cornea, making them more likely to cause corneal edema and epithelial lesions.
The corneal curvature radius of infants should be measured under anesthesia, along with refractive power. Contact lenses can be fitted as early as 6 days after cataract surgery. The refractive power decreases with age. Generally, for infants under 1 month, it is +35D; at 1 month, +30D; at 2 months, +25D; and at 3 months, +20D. Regular refractive examinations are necessary: weekly for infants under 2 months, twice a month for those under 1 month, once a month for those under 2 months, and the intervals can be extended thereafter. Either hydrophilic soft lenses or hard lenses can be used, but high plus-power lenses are often unsuitable. School-aged children should have two pairs of lenses: one for distance vision and one for reading.
The improvement of vision after unilateral congenital cataract surgery largely depends on parental cooperation and patience. Infants under 1 year of age blink less frequently, making lens loss more likely. Children aged 2–6 years are often uncooperative, requiring frequent lens replacements. When initiating contact lens use for unilateral cataracts, the healthy eye should be strictly patched. If eccentric fixation persists after 6 months of patching, it indicates irreversible amblyopia, and patching may be discontinued.
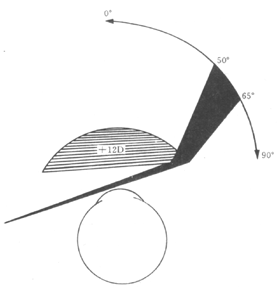
Figure 1 Peripheral prism effect of high hyperopia lenses
In summary, to restore vision in congenital cataracts and reduce the incidence of amblyopia and blindness, the importance of early surgery should be emphasized, along with active optical correction measures postoperatively.
Many patients with congenital cataracts are often associated with other eye diseases or abnormalities, and the presence of these complications further exacerbates visual impairment. Therefore, when diagnosing and treating congenital cataracts, it is important to pay attention to the existence of these complications in order to take appropriate therapeutic measures.
1. Strabismus: More than half of patients with unilateral cataracts and less than half of those with bilateral cataracts are accompanied by strabismus. Due to lens opacity or changes in refractive power in one eye, visual acuity declines; or the imbalance in visual acuity caused by differing degrees of lens opacity in both eyes disrupts the fusion mechanism, gradually leading to strabismus. Additionally, eyes affected by congenital cataracts may have certain anatomical abnormalities (such as microphthalmia) and intraocular diseases, which can also cause and progressively worsen strabismus. Some systemic disorders may present with congenital cataracts combined with strabismus, such as Lowe syndrome, Stickler syndrome, neonatal hemolytic disease, and certain chromosomal abnormality syndromes.
2. Nystagmus: Because congenital cataracts impair vision, patients may develop pendular or searching nystagmus, known as secondary nystagmus, which may lessen or disappear after cataract surgery. If nystagmus persists postoperatively, it will inevitably affect visual recovery. Congenital cataracts combined with nystagmus can also be seen in certain systemic diseases, such as mandibulofacial dysostosis syndrome, deletion of the long arm of chromosome 21, and Marinesco-Sjögren syndrome.
3. Congenital Microphthalmia: For patients with congenital cataracts combined with congenital microphthalmia, visual recovery is unlikely to be ideal, even after cataract surgery, as the improvement in vision is limited. The presence of congenital microphthalmia is unrelated to the type of congenital cataract; it may result from changes in eye size during abnormal lens development leading to lens opacity, often with a genetic component. In addition to microphthalmia, defects in certain intraocular tissues (such as the iris or choroid) may also occur. Congenital cataracts combined with microphthalmia can also be observed in certain systemic diseases, such as Norrie disease, Gruber disease, and some chromosomal aberration syndromes.
4. Retinal and Choroidal Lesions: A small number of patients with congenital cataracts may also have myopic choroidal retinopathy, retinitis pigmentosa, Leber congenital amaurosis, or macular dystrophy.
5. Hypoplasia of the Iris Dilator Muscle: The pupil does not dilate easily after the application of mydriatic drops, which poses certain difficulties in the examination and surgery of cataract patients.
6. Others: In addition to the more common complications mentioned above, patients may also have lens dislocation, lens defects, congenital aniridia, congenital iris and/or choroidal defects, persistent pupillary membrane, megalocornea, keratoconus, persistent hyperplastic primary vitreous, and other conditions.
Congenital white internal visual obstruction has different disease causes and varied clinical manifestations. To establish a clear diagnosis, the following laboratory tests should be completed.
1. Congenital white internal visual obstruction combined with other systemic malformations may indicate chromosomal disorders. Therefore, chromosomal karyotyping and banding analysis should be performed.
2. For diabetes and neonatal hypoglycemia, blood glucose, urine glucose, and ketone bodies should be tested.
3. For nephropathy combined with congenital white internal visual obstruction, routine urinalysis and urine amino acids should be examined to confirm diagnoses such as Lowe syndrome and Alport syndrome.
4. For phenylketonuria, positive urine phenylpyruvic acid and positive ferric chloride test in urine are indicative.
5. For hypoparathyroidism, serum calcium is decreased while serum phosphorus is elevated. If serum calcium is below 1.92 mmol/L, hypocalcemic white internal visual obstruction may occur.
6. For galactosemia, in addition to screening for galactosuria, galactose-1-phosphate uridyltransferase and galactokinase should be tested.
7. For homocystinuria, qualitative testing for homocystine in urine should be performed. A positive sodium borohydride test confirms the diagnosis.
8. Amino acid measurement using an automated amino acid analyzer to determine blood amino acid levels can diagnose certain metabolic disorders associated with congenital white internal visual obstruction, such as homocystinuria and tyrosinemia.
9. For rubella syndrome, maternal serum during the acute phase or convalescence stage should be tested for rubella virus antibodies. A titer four times higher than normal is considered a positive result.
Since congenital white internal visual obstruction may also be associated with other eye diseases, in addition to the necessary laboratory tests mentioned above, B-scan ultrasound, electroretinography, and visual evoked potential tests should be conducted to predict postoperative visual recovery after white internal visual obstruction surgery.


