| disease | Hepatic Encephalopathy |
| alias | Hepatic Unconsciousness, HE, Hepatic EncepHalopathy |
Hepatic encephalopathy (HE), previously known as hepatic coma, is a syndrome of central nervous system dysfunction caused by severe liver disease and based on metabolic disturbances. Its main clinical manifestations are consciousness disorders, behavioral abnormalities, and unconsciousness. Porto-systemic encephalopathy (PSE) emphasizes portal hypertension, where collateral circulation exists between the portal vein and the vena cava, allowing a large amount of portal venous blood to bypass the liver and enter the systemic circulation, which is the main mechanism of encephalopathy. Subclinical or latent hepatic encephalopathy refers to hepatic encephalopathy without obvious clinical manifestations and generation and transformation abnormalities, which can only be diagnosed through detailed intellectual tests and/or electrophysiological detection.
bubble_chart Etiology
Most hepatic encephalopathy is caused by various types of cirrhosis (post-hepatitis cirrhosis being the most common), including surgical portosystemic shunt procedures for treating portal hypertension in cirrhosis. If subclinical hepatic encephalopathy is also included, the incidence of hepatic encephalopathy in cirrhosis patients can reach 70%. A small portion of encephalopathy cases are seen in severe viral hepatitis, toxic hepatitis, and the acute or fulminant liver failure stages of drug-induced liver disease. Even rarer disease causes include primary liver cancer, acute fatty liver during pregnancy, and severe biliary tract infections.
Hepatic encephalopathy, especially portosystemic encephalopathy, often has obvious triggers, such as upper gastrointestinal bleeding, excessive potassium loss due to diuretics, ascites drainage, high-protein diet, sedatives, anesthetics, constipation, uremia, surgical procedures, and infections.
The mechanism of hepatic encephalopathy is not yet fully understood. It is generally believed that the pathophysiological basis of hepatic encephalopathy is the failure of liver cell function and the presence of surgical or naturally formed collateral shunts between the portal and systemic circulation. Mainly, many toxic metabolites from the intestines are not detoxified and cleared by the liver, entering the systemic circulation through collateral vessels, crossing the blood-brain barrier, and reaching the brain, causing cerebral dysfunction. The metabolic disturbances in hepatic encephalopathy are multifaceted, and the occurrence of encephalopathy may be the result of multiple factors, but metabolic disorders of nitrogenous substances including proteins, amino acids, ammonia, and thiols, and the accumulation of inhibitory neurotransmitters may play a major role. Disorders in glucose, water, and electrolyte metabolism, as well as hypoxia, can interfere with brain energy metabolism and exacerbate encephalopathy. Abnormal fat metabolism, especially the increase in short-chain fatty acids, also plays an important role. Additionally, increased brain sensitivity in patients with chronic liver disease is also a significant factor. There are many hypotheses regarding the mechanism of hepatic encephalopathy, with the ammonia intoxication theory being the most studied and substantiated.
(1) Ammonia intoxication: Ammonia intoxication caused by ammonia metabolism disorder is an important mechanism of hepatic encephalopathy, especially portal-systemic encephalopathy, and encephalopathy related to ammonia intoxication is also called nitrogenous encephalopathy. 1. Formation and metabolism of ammonia: Blood ammonia mainly comes from the ammonia generated in the intestines, kidneys, and skeletal muscles, but the gastrointestinal tract is the main portal for ammonia to enter the body. Normally, the gastrointestinal tract can produce 4g of ammonia daily, most of which is produced by the decomposition of urea diffused into the intestines from the blood circulation by bacterial urease, and a small part is produced by the decomposition of proteins in food by bacterial amino acid oxidase. The absorption of ammonia in the intestines mainly occurs in the form of non-ionic ammonia (NH3) diffusing into the intestinal mucosa, and its absorption rate is much higher than that of ionic ammonia (NH4+). Free NH3 is toxic and can cross the blood-brain barrier; NH4+ exists in the form of salts, is relatively non-toxic, and cannot cross the blood-brain barrier. The mutual conversion of NH3 and NH4+ is influenced by changes in pH gradient. As shown in the reaction formula 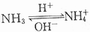 , when the colonic pH > 6, NH3 diffuses into the blood in large amounts; when pH < 6, NH4 transfers from the blood to the intestinal cavity and is excreted with feces. The kidney produces ammonia by decomposing glutamine in the renal blood flow into ammonia by glutaminase in the renal tubular epithelial cells. When the renal tubular filtrate is alkaline, a large amount of NH3 is absorbed into the renal vein, increasing blood ammonia; when it is acidic, ammonia enters the renal tubular lumen in large amounts and combines with acid, and is excreted from the body in the form of ammonium salts (such as NH4Cl), which is an important way for the kidney to excrete strong acids. In addition, skeletal muscles and myocardium can also produce ammonia during exercise.
, when the colonic pH > 6, NH3 diffuses into the blood in large amounts; when pH < 6, NH4 transfers from the blood to the intestinal cavity and is excreted with feces. The kidney produces ammonia by decomposing glutamine in the renal blood flow into ammonia by glutaminase in the renal tubular epithelial cells. When the renal tubular filtrate is alkaline, a large amount of NH3 is absorbed into the renal vein, increasing blood ammonia; when it is acidic, ammonia enters the renal tubular lumen in large amounts and combines with acid, and is excreted from the body in the form of ammonium salts (such as NH4Cl), which is an important way for the kidney to excrete strong acids. In addition, skeletal muscles and myocardium can also produce ammonia during exercise.
The main pathways for the body to eliminate blood ammonia are: ① Urea synthesis - the majority of ammonia from the intestines is converted into urea in the liver through the ornithine metabolic cycle; ② Under the energy supply of adenosine triphosphate (ATP), tissues such as the brain, liver, and kidney utilize and consume ammonia to synthesize glutamate and glutamine (α-ketoglutarate + NH3
→ glutamate, glutamate + NH3 → glutamine); ③ The kidney is the primary site for ammonia excretion, in addition to excreting a large amount of urea, it also eliminates a significant amount of ammonia in the form of NH4+ while excreting acid; ④ When blood ammonia levels are excessively high, a small amount can be exhaled through the lungs.2. Causes of elevated blood ammonia in hepatic encephalopathy The increase in blood ammonia is mainly due to excessive production and/or insufficient metabolic clearance. Excessive production of blood ammonia can be exogenous, such as excessive intake of nitrogen-containing food or drugs from outside the body, which are converted into ammonia in the intestines; it can also be endogenous, such as in cases of prerenal and renal azotemia, where a large amount of urea in the blood diffuses into the intestinal lumen, is converted into ammonia, and then enters the bloodstream. After gastrointestinal bleeding, the blood remaining in the intestines decomposes into ammonia, which is not from outside the body and should be considered endogenous, but the process of ammonia production is similar to that of ingesting nitrogen-containing foods. In summary, during liver failure, the liver's ability to synthesize ammonia into urea is reduced, and when portosystemic shunting is present, intestinal ammonia bypasses the liver's detoxification process and directly enters the systemic circulation, leading to elevated blood ammonia.
3. Factors affecting ammonia toxicity Many factors that induce hepatic encephalopathy can affect the amount of ammonia entering the brain tissue and/or alter the brain tissue's sensitivity to ammonia.
(1) Hypokalemic alkalosis: Reduced food intake, vomiting, diarrhea, potassium loss from diuresis, ascites drainage, and secondary hyperaldosteronism can all lead to hypokalemia. Hypokalemia causes acid-base imbalance, thereby altering the distribution of ammonia inside and outside the cells. When potassium is lost from the extracellular fluid, it is replaced by potassium moving out of the cells, and the displaced potassium is exchanged with sodium and hydrogen from the extracellular fluid, thus reducing [H+] in the extracellular fluid, which facilitates the entry of NH3
into brain cells to produce toxic effects. Moreover, the excretion of potassium and hydrogen by the kidneys is negatively correlated; in hypokalemia, the excretion of potassium decreases while the excretion of hydrogen ions increases, leading to metabolic alkalosis, which promotes NH3 to cross the blood-brain barrier and enter cells to cause harm. Most patients with portosystemic encephalopathy have elevated blood ammonia, and consciousness can return to normal after blood ammonia is reduced; many cases of fulminant liver failure remain deeply unconscious but have normal blood ammonia. Additionally, cirrhotic patients who develop encephalopathy due to the use of sedatives, hypnotics, or anesthetics may also have normal or slightly elevated blood ammonia, which are considered non-nitrogenous encephalopathy, accounting for about one-third of all encephalopathy cases.(2) Excessive intake of nitrogen-containing foods or drugs, or upper gastrointestinal bleeding (approximately 20g of protein per 100ml of blood) increases ammonia production in the intestines.
(3) Hypovolemia and hypoxia: Seen in upper gastrointestinal bleeding, massive ascites drainage, diuresis, etc. Shock and hypoxia can lead to prerenal azotemia, increasing blood ammonia. Hypoxia in brain cells can reduce the brain's tolerance to ammonia toxicity.
(4) Constipation: Prolongs the contact time of ammonia, amines, and other toxic derivatives with the colonic mucosa, facilitating the absorption of toxins.
(5) Infection: Increases tissue catabolism, thereby increasing ammonia production; dehydration can exacerbate prerenal azotemia; hypoxia and high fever increase the toxicity of ammonia.
(6) Hypoglycemia: Glucose is an important fuel for energy production in the brain; during hypoglycemia, energy decreases, ammonia removal activities in the brain stagnate, and the toxicity of ammonia increases.
(7) Others: Sedatives and hypnotics can directly inhibit the brain and respiratory centers, causing hypoxia. Anesthesia and surgery increase the functional burden on the liver, brain, and kidneys.
4. Toxic effects of ammonia on the central nervous system. Brain cells are extremely sensitive to ammonia. In normal individuals, skeletal muscle, liver, and brain tissue can uptake excess ammonia from the blood (accounting for 50%, 24%, and 7.5% respectively). In cirrhosis, ammonia uptake is often reduced due to muscle wasting, and liver uptake is also reduced due to portosystemic shunting, resulting in a greater ammonia load on the brain. It is generally believed that the toxic effect of ammonia on the brain is to interfere with cerebral energy metabolism, leading to a decrease in the concentration of high-energy phosphate compounds. High blood ammonia levels may inhibit the activity of pyruvate dehydrogenase, thereby affecting the generation of acetyl-CoA and interfering with the tricarboxylic acid cycle in the brain. On the other hand, during the detoxification process of ammonia in the brain, ammonia combines with α-ketoglutarate to form glutamate, and glutamate combines with ammonia to form glutamine. These reactions consume large amounts of coenzymes, ATP, α-ketoglutarate, and glutamate, and generate large amounts of glutamine. α-Ketoglutarate is an important intermediate in the tricarboxylic acid cycle, and its deficiency leads to insufficient energy supply to brain cells, preventing them from maintaining normal function. Glutamate is an important excitatory neurotransmitter in the brain, and its deficiency increases brain inhibition.
(II) Synergistic Toxic Effects of Ammonia, Mercaptans, and Short-Chain Fatty Acids Methyl mercaptan is a product of methionine metabolism by bacteria in the gastrointestinal tract. Both methyl mercaptan and its derivative, dimethyl sulfoxide, can cause confusion, disorientation, lethargy, and unconsciousness in experimental animals. The mechanism of hepatic encephalopathy in cirrhotic patients after consuming methionine may be related to these two metabolites. The characteristic liver odor may be due to the volatilization of methyl mercaptan and dimethyl disulfide. In patients with severe liver disease, blood concentrations of methyl mercaptan are elevated, and this elevation is more pronounced in those with encephalopathy. Short-chain fatty acids (mainly valeric acid, caproic acid, and caprylic acid) are formed by bacterial decomposition of long-chain fatty acids and can induce experimental hepatic encephalopathy. These acids are also significantly elevated in the plasma and cerebrospinal fluid of patients with hepatic encephalopathy.
In experimental animals with liver failure, the use of any one of the three toxic substances—ammonia, mercaptans, or short-chain fatty acids—in small doses is insufficient to induce hepatic encephalopathy. However, when used in combination, even without increasing the dose, they can cause brain symptoms. Therefore, some scholars propose that the synergistic toxic effects of ammonia, mercaptans, and short-chain fatty acids on the central nervous system may play an important role in the pathogenesis of hepatic encephalopathy.
(III) False Neurotransmitters The conduction of nerve impulses is mediated by neurotransmitters. Neurotransmitters are divided into excitatory and inhibitory types, which normally maintain a physiological balance. Excitatory neurotransmitters include dopamine and norepinephrine from catecholamines, acetylcholine, glutamate, and aspartate. Inhibitory neurotransmitters are only formed in the brain.
Aromatic amino acids in food, such as tyrosine and phenylalanine, are converted by bacterial decarboxylase into tyramine and phenylethylamine, respectively. Normally, these amines are broken down and cleared by monoamine oxidase in the liver. In liver failure, this clearance is impaired, allowing these amines to enter the brain. Neijing Under the action of β-hydroxylase, they are converted into 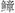 amines (β-hydroxyltyramine) and phenylethanolamine. The chemical structures of these two substances are similar to the normal neurotransmitter norepinephrine, but they cannot transmit nerve impulses or do so weakly, hence they are called false neurotransmitters. When false neurotransmitters are taken up by brain cells and replace normal neurotransmitters in synapses, nerve conduction is impaired, and excitatory impulses cannot be transmitted normally to the cerebral cortex, leading to abnormal inhibition, consciousness disorders, and unconsciousness.
amines (β-hydroxyltyramine) and phenylethanolamine. The chemical structures of these two substances are similar to the normal neurotransmitter norepinephrine, but they cannot transmit nerve impulses or do so weakly, hence they are called false neurotransmitters. When false neurotransmitters are taken up by brain cells and replace normal neurotransmitters in synapses, nerve conduction is impaired, and excitatory impulses cannot be transmitted normally to the cerebral cortex, leading to abnormal inhibition, consciousness disorders, and unconsciousness.
To date, the theory of these false neurotransmitters has not been fully confirmed.
(IV) GABA/Bz Receptor γ-Aminobutyric acid (GABA) is the major inhibitory neurotransmitter in the mammalian brain, produced by intestinal bacteria. In cases of portosystemic shunting and liver failure, GABA can bypass the liver and enter the systemic circulation. Recent studies in animal models of fulminant liver failure and hepatic encephalopathy have found increased blood concentrations of GABA and increased permeability of the blood-brain barrier. The number of GABA receptors on postsynaptic neurons in the brain is significantly increased. These receptors can not only bind to GABA but also to barbiturates and benzodiazepines (BZs) at different sites on the receptor surface, hence they are called GABA/BZ complex receptors. Whether GABA or any of the aforementioned drugs bind to the receptor, they promote chloride ion conduction into the postsynaptic neuron, leading to inhibition of nerve conduction. The visual evoked potentials (VEP) recorded by instruments in this state are the same as those in animal models of encephalopathy induced by galactosamine. The plasma GABA concentration in patients with hepatic encephalopathy parallels the degree of encephalopathy. A few patients have shown symptom improvement and normalization of VEP after treatment with GABA receptor antagonists or benzodiazepine receptor antagonists, further supporting the idea that hepatic encephalopathy is caused by an increase in the inhibitory neurotransmitter GABA.
(5) Imbalance in Amino Acid Metabolism: Plasma amino acid measurements reveal that in decompensated liver cirrhosis patients, plasma levels of aromatic amino acids (such as phenylalanine, tyrosine, and tryptophan) increase while branched-chain amino acids (such as valine, leucine, and isoleucine) decrease, indicating an imbalance in the metabolism of these two groups of amino acids. In healthy individuals, aromatic amino acids are metabolized and broken down in the liver. In cases of liver failure, this breakdown decreases, leading to increased blood concentrations. Normally, branched-chain amino acids are primarily metabolized in skeletal muscles rather than the liver, but insulin facilitates the entry of these amino acids into muscles. In liver failure, the inactivation of insulin in the liver is reduced, leading to increased blood insulin levels, which in turn promotes the entry of branched-chain amino acids into muscle tissues, resulting in decreased blood concentrations. This ultimately reduces the molar ratio of branched-chain to aromatic amino acids from the normal range of 3-3.5 to 1 or lower. These two groups of amino acids compete and exclude each other as they pass through the blood-brain barrier into the brain, exchanging with glutamine. A decrease in branched-chain amino acids leads to an increase in aromatic amino acids entering the brain, which can further form false neurotransmitters as previously mentioned. In liver cirrhosis patients, due to impaired liver metabolism and reduced plasma albumin levels, serum free tryptophan increases. The increased tryptophan in the brain can be converted into serotonin, an inhibitory neurotransmitter for certain central nervous system neurons, which antagonizes norepinephrine and may also be related to unconsciousness. Arginine, glutamic acid, and aspartic acid, or their derivatives, have been shown to reverse experimental hepatic encephalopathy caused by ammonia toxicity and have awakening effects in liver cirrhosis patients with unconsciousness.
bubble_chart Pathological Changes
In patients with hepatic encephalopathy caused by acute liver failure, the brain often shows no significant anatomical abnormalities, but 38-50% have cerebral edema, which may be a secondary change of the condition. In chronic hepatic encephalopathy patients, there may be protoplasmic astrocytic hypertrophy and proliferation in the gray matter of the cerebrum and cerebellum, as well as in subcortical tissues. In cases with a longer course, the cerebral cortex may thin, with neurons and nerve fibers disappearing, and patchy necrosis may occur in the deep layers of the cortex, potentially even affecting the cerebellum and basal regions.
bubble_chart Clinical Manifestations
The clinical manifestations of hepatic encephalopathy often vary greatly depending on the nature of the underlying liver disease, the severity and urgency of liver cell damage, and the different inducing factors. Acute hepatic encephalopathy is commonly seen in fulminant hepatitis, with massive liver cell necrosis and acute liver failure, and the inducing factors are not obvious. Patients may enter unconsciousness within a few days of onset and progress to death, and there may be no prodromal symptoms before unconsciousness. Chronic hepatic encephalopathy is mostly portal-systemic encephalopathy, caused by extensive portal-systemic collateral circulation and chronic liver failure, commonly seen in patients with cirrhosis and/or after portacaval shunt surgery, with chronic recurrent stupor and unconsciousness as prominent manifestations. Common inducing factors include a high-protein diet, upper gastrointestinal bleeding, infection, ascites drainage, and massive potassium-losing diuresis. Hepatic encephalopathy seen in the terminal stage of cirrhosis has a slow onset, with unconsciousness gradually deepening until death.
To observe the dynamic changes of encephalopathy, facilitate early diagnosis and management, and analyze therapeutic efficacy, hepatic encephalopathy is generally divided into four stages based on the degree of consciousness impairment, neurological manifestations, and electroencephalogram (EEG) changes, ranging from mild mental changes to deep unconsciousness:
Initial stage [first stage] (prodromal stage) Grade I: Personality changes and behavioral abnormalities, such as euphoria or apathy, disheveled appearance, or inappropriate urination and defecation. Responses are still accurate, but speech is slurred and slow. Flapping tremor (asterixis), also known as liver tremor, may be present: When the patient is asked to extend both arms with the elbows fixed and the palms dorsiflexed with fingers spread, the hands may deviate outward, with rapid and irregular flapping movements at the metacarpophalangeal joints, wrist joints, and even the elbows and shoulders. If the patient is asked to grip the doctor's hand for one minute, the doctor can feel the patient's tremor. The EEG is mostly normal. This stage lasts for days or weeks, and sometimes the symptoms are not obvious and easily overlooked.
Intermediate stage [second stage] (pre-unconsciousness stage): Characterized mainly by confusion, sleep disturbances, and behavioral abnormalities. Symptoms from the initial stage [first stage] worsen, with impaired orientation and comprehension, confusion about time, place, and person, and inability to perform simple calculations or intellectual tasks (such as building blocks or arranging matchsticks into a star). Slurred speech, writing difficulties, and abnormal behavior are also common. Sleep inversion, with daytime sleepiness and nighttime wakefulness, is frequent, and hallucinations, fear, and mania may occur, leading to misdiagnosis as a general psychiatric disorder. This stage is marked by obvious neurological signs, such as hyperreflexia, increased muscle tone, ankle clonus, and positive Babinski sign. Flapping tremor persists, and the EEG shows characteristic abnormalities. Involuntary movements and ataxia may appear.
Late stage [third stage] (stupor stage): Characterized mainly by stupor and confusion, with various neurological signs persisting or worsening. The patient is in a stuporous state most of the time but can be aroused. When awake, they can respond to questions but often exhibit confusion and hallucinations. Flapping tremor can still be elicited. Muscle tone increases, and passive limb movements often encounter resistance. Pyramidal signs are usually positive, and the EEG shows abnormal waveforms.
Fourth stage (unconsciousness stage): Complete loss of consciousness, unarousable. In light unconsciousness, there is still a response to painful stimuli and uncomfortable positions, with hyperreflexia and increased muscle tone. Due to the patient's inability to cooperate, flapping tremor cannot be elicited. In deep unconsciousness, all reflexes disappear, muscle tone decreases, pupils are often dilated, and paroxysmal convulsions, ankle clonus, and hyperventilation may occur. The EEG shows significant abnormalities.
The boundaries between the above stages are not very clear, and clinical manifestations of the late stage [third stage] may overlap. The severity may progress or regress with disease progression or treatment. A few patients with chronic hepatic encephalopathy may exhibit intellectual decline, ataxia, positive pyramidal signs, or paraplegia due to organic damage in different parts of the central nervous system. These manifestations may be temporary or become permanent.
Patients with subclinical or latent hepatic encephalopathy are considered healthy individuals with no clinical manifestations and participate in normal social activities. There is a risk of traffic accidents when driving various vehicles, which has garnered significant attention in Western countries in recent years. Some have suggested that subclinical hepatic encephalopathy should be classified as stage 0 in the clinical staging system.
Severe hepatic encephalopathy with significant liver function impairment often presents with obvious jaundice, bleeding tendency, and hepatic odor, and is prone to complications such as various infections, hepatorenal syndrome, and cerebral edema, making the clinical manifestations more complex.
bubble_chart Auxiliary Examination
(1) Blood Ammonia The normal fasting venous blood ammonia level in healthy individuals is 40-70 μg/dl, while the stirred pulse blood ammonia content is 0.5-2 times that of venous blood ammonia. Fasting stirred pulse blood ammonia is relatively stable and reliable. Patients with chronic hepatic encephalopathy, especially those with portosystemic encephalopathy, often have elevated blood ammonia levels. In encephalopathy caused by acute liver failure, blood ammonia levels are usually normal.
(2) Electroencephalogram (EEG) Examination EEG not only has diagnostic value but also carries certain prognostic significance. The typical changes include a slowing of rhythm, primarily showing generalized theta waves of 4-7 Hz, and in some cases, delta waves of 1-3 Hz. During unconsciousness, high-amplitude delta waves appear symmetrically on both sides.
(3) Evoked Potentials These are externally recordable potentials generated by synchronous discharge responses in the brain's neural network after various external stimuli are transmitted to sensory receptors. Depending on the sensory organ stimulated, they are classified as visual evoked potentials (VEP), auditory evoked potentials (AEP), and somatosensory evoked potentials (SEP). It has been observed that the evoked potentials recorded from experimental rats or rabbits with hepatic encephalopathy show specific changes corresponding to the severity of the condition. This technique was later applied to study patients with hepatic encephalopathy. Initially, it was believed that VEP could provide an objective and accurate diagnosis of hepatic encephalopathy at various stages, including subclinical encephalopathy, with sensitivity surpassing any other method. However, recent studies suggest that VEP examinations vary too much among individuals and over time, lacking specificity and sensitivity, and are less effective than simple psychological or intelligence tests.
(4) Simple Intelligence Tests Currently, intelligence tests are considered most useful for diagnosing early hepatic encephalopathy, including subclinical encephalopathy. The test content includes writing, word construction, drawing, block building, and constructing a five-pointed star with matchsticks. The most commonly used routine test is the number connection test, as its results are easy to quantify and facilitate follow-up.
The main diagnostic criteria for hepatic encephalopathy are: ① severe liver disease and/or extensive portosystemic collateral circulation; ② mental confusion, lethargy, or unconsciousness; ③ precipitating factors of hepatic encephalopathy; ④ significant liver dysfunction or elevated blood ammonia levels. Flapping tremor (asterixis) and typical electroencephalogram (EEG) changes have important reference value.
Routine simple intelligence tests for patients with cirrhosis can detect subclinical hepatic encephalopathy.
Hepatic encephalopathy presenting solely with psychiatric symptoms is easily misdiagnosed as a psychiatric disorder. Therefore, when encountering patients with mental confusion, the possibility of hepatic encephalopathy should be considered. Hepatic unconsciousness should also be differentiated from other conditions that can cause unconsciousness, such as diabetes, hypoglycemia, uremia, cerebrovascular accidents, brain infections, and sedative overdose. Further inquiry into the history of liver disease, examination of liver and spleen size, liver function tests, blood ammonia levels, and EEG will aid in diagnosis and differential diagnosis.
bubble_chart Treatment Measures
Hepatic encephalopathy currently has no specific treatment, and comprehensive measures should be taken:
(1) Eliminating precipitating factors. Certain factors can induce or exacerbate hepatic encephalopathy. In cirrhosis, the half-life of drugs in the body is prolonged, clearance is reduced, and the sensitivity of the brain in encephalopathy patients is increased. Most cannot tolerate drugs such as anesthetics, analgesics, hypnotics, and sedatives. Improper use can lead to drowsiness, even unconsciousness. When patients are agitated or have spasms, morphine and its derivatives, paraldehyde, chloral hydrate, pethidine, and fast-acting barbiturates are contraindicated. Diazepam and scopolamine can be used in reduced doses (half or one-third of the usual dose) with reduced frequency. Antihistamines such as phenergan and chlorpheniramine can sometimes be used as substitutes for sedatives. It is essential to promptly control infections and upper gastrointestinal bleeding, avoid rapid and massive potassium-depleting diuresis and ascites drainage. Attention should be paid to correcting typical edema, electrolyte, and acid-base imbalances.
(2) Reducing the production and absorption of intestinal internal toxins
1. Diet. Protein should be avoided in the first few days. Provide 1200-1600 calories per day and sufficient vitamins, with carbohydrates as the main food. For those who cannot eat due to unconsciousness, feeding can be done via a nasogastric tube. Fat can delay gastric emptying and should be used sparingly. The best nasogastric feeding solution is 25% sucrose or glucose solution, producing 1 calorie per ml, and 3-6g of essential amino acids can be given daily. If the stomach cannot empty, nasogastric feeding should be stopped and replaced with deep venous catheter infusion of 25% glucose solution to maintain nutrition. During massive glucose infusion, be vigilant for hypokalemia, heart failure, and cerebral edema. After regaining consciousness, protein can be gradually increased to 40-60g/day. The tendency of different proteins to cause unconsciousness varies; generally, meat protein has the greatest effect on encephalopathy, followed by milk protein, and plant protein the least. Therefore, correcting the patient's negative nitrogen balance is best achieved with plant protein. Plant protein contains less methionine and aromatic amino acids, more branched-chain amino acids, and can increase fecal nitrogen excretion. Additionally, plant protein contains non-absorbable fibers, which are fermented by intestinal bacteria to produce acids beneficial for ammonia excretion and facilitate bowel movements, making it suitable for patients with hepatic encephalopathy.
2. Enema or catharsis. To clear intestinal food residues, blood, or other nitrogenous substances, physiological saline or weakly acidic solution (e.g., dilute acetic acid solution) can be used for enema, or 25% magnesium sulfate 30-60ml can be given orally or via nasogastric tube for catharsis. For acute portal-systemic encephalopathy unconscious patients, lactulose 500ml mixed with 500ml water for enema is particularly useful as an initial treatment.
3. Inhibiting bacterial growth. Oral neomycin 2-4g/day or paromomycin, kanamycin, or ampicillin can be effective. A few patients on long-term neomycin may experience hearing or renal function impairment, so neomycin should not be used for more than a month. Oral metronidazole 0.2g, four times daily, is equally effective as neomycin and suitable for patients with renal insufficiency.
Lactulose (β-galactosidofructose), when taken orally, is broken down by bacteria in the colon into lactic acid and acetic acid, which acidifies the intestinal lumen, thereby reducing the formation and absorption of ammonia. For patients who are intolerant to neomycin or require long-term treatment, lactulose or lactitol is the drug of choice. Lactulose is available in syrup and powder forms, with a daily dose of 30-100ml or 30-100g, divided into three oral doses. Starting with a small dose, it is adjusted to achieve 2-3 bowel movements per day, with a stool pH of 5-6. Side effects include bloating, colicky abdominal pain, nausea, and vomiting. Lactitol (β-galactosido-sorbitol) is a disaccharide similar to lactulose, available in tablet or syrup form, easy to store, with the same metabolic pathway and efficacy as lactulose, and a daily dose of 30g, divided into three oral doses. Recent studies have found that lactose, when fermented by bacteria in the colon of individuals with lactase deficiency, also lowers stool pH and reduces ammonia levels, making it as effective as lactulose in treating hepatic encephalopathy, but at a lower cost.
(3) Promote the metabolic elimination of toxic substances and correct the disorder of amino acid metabolism.
1. Ammonia-lowering drugs: ① Potassium glutamate (6.3g/20ml per ampoule, containing 34mmol of potassium) and sodium glutamate (5.75g/20ml per ampoule, containing 34mmol of sodium), 4 ampoules each time, added to glucose solution for intravenous drip, once or twice daily. The ratio of potassium glutamate to sodium glutamate depends on the serum potassium and sodium concentrations and the condition of the patient. Use less potassium in cases of oliguria and use sodium cautiously in cases of significant ascites and edema. ② Arginine 10-20g added to glucose solution for intravenous drip once daily. This drug promotes urea synthesis and is acidic, suitable for patients with high blood pH. Ammonia-lowering Yaodui is more effective for chronic recurrent portosystemic encephalopathy but ineffective for acute hepatic unconsciousness caused by severe hepatitis. ③ Sodium benzoate can combine with residual nitrogen substances in the intestine such as glycine or glutamine to form hippuric acid, which is excreted by the kidneys, thus reducing blood ammonia. Its effect on acute portosystemic encephalopathy is comparable to lactulose. The dose is 5g orally twice daily. ④ Phenylacetic acid combines with intestinal glutamine to form non-toxic hippuric acid, which is excreted by the kidneys, also reducing blood ammonia concentration. ⑤ Ornithine-α-ketoglutarate and ornithine aspartate both have significant ammonia-lowering effects.
2. Branched-chain amino acids: Oral or intravenous infusion of amino acid mixtures mainly composed of branched-chain amino acids can theoretically correct the imbalance of amino acid metabolism and inhibit the formation of false vitality neurotransmitters in the brain, but the efficacy for portosystemic encephalopathy is still controversial. Branched-chain amino acids have a lesser unconsciousness-inducing effect compared to general dietary proteins. If patients cannot tolerate protein foods, intake of sufficient amounts of branched-chain amino acid-rich mixtures is effective and safe for restoring positive nitrogen balance.
3. GABA/BZ complex receptor antagonists: The GABA receptor antagonist bicuculline and the weak tranquilizer receptor antagonist flumazenil are available. The dose of flumazenil has a wide range. It has been reported that intravenous infusion of 15mg of flumazenil over 3 hours significantly improved symptoms and somatosensory evoked potentials (SEP) in 45% of patients with fulminant hepatic failure encephalopathy and 78% of patients with cirrhosis, but symptoms recurred several hours after discontinuation. Another group reported a dose of 0.2mg intravenous injection of flumazenil. If there is no improvement in EEG after 3 minutes, the dose is increased to 0.4mg, then 0.8mg, 1-2mg, with a maximum total dose of 9.6mg in one case, and 71% of 14 patients showed improvement. The dose used in our hospital is 0.5mg added to 10ml of 0.9% saline, injected over 5 minutes, followed by 1.0mg added to 250ml of saline, dripped over 30 minutes, which greatly improves symptoms in patients with cirrhosis and hepatic encephalopathy.
(4) Liver transplantation: For many chronic liver diseases that currently have no other satisfactory treatment methods to reverse, liver transplantation is a recognized effective treatment. Due to improvements and standardization of the transplantation procedure, advances in liver preservation methods and surgical techniques, and the use of low-toxicity immunosuppressants for anti-rejection, the survival rate of patients after transplantation has significantly improved (refer to the chapter on liver transplantation).
(5) Other symptomatic treatments
1. Correct typical edema, electrolyte, and acid-base balance disorders: The total daily fluid intake should not exceed 2500ml. Fluid intake in patients with cirrhosis and ascites should be controlled (generally about urine output plus 1000ml) to avoid blood dilution, hyponatremia, and worsening unconsciousness. Correct potassium deficiency and alkalosis promptly; potassium chloride is supplemented for potassium deficiency, and arginine salt solution is used for intravenous drip in cases of alkalosis.
2. Protect brain cell function: Use an ice cap to lower intracranial temperature to reduce energy consumption and protect brain cell function.
3. Maintain airway patency. For those in deep unconsciousness, perform a tracheotomy and administer oxygen.
4. Prevention and treatment of cerebral edema: Intravenous infusion of hypertonic glucose, mannitol, and other dehydrating agents to prevent and treat cerebral edema.
5. Prevention of bleeding and shock: For those with a tendency to bleed, intravenous infusion of vitamin K1 or fresh blood transfusion may be administered to correct shock, hypoxia, and prerenal uremia.
6. Peritoneal or renal dialysis: If azotemia is the cause of hepatic encephalopathy, peritoneal or hemodialysis may be useful.
The prognosis is better for those with clear and easily eliminable causes (such as bleeding, potassium deficiency, etc.). Patients with better liver function, those who have undergone shunt surgery, and those with portosystemic encephalopathy caused by high protein intake have a better prognosis. Patients with ascites, jaundice, and a tendency to bleed indicate very poor liver function and a poor prognosis. The prognosis for hepatic encephalopathy caused by fulminant liver failure is the worst.
Actively prevent and treat liver disease. Patients with liver disease should avoid all factors that may induce hepatic encephalopathy. Closely monitor patients with liver disease, promptly identify the prodromal and pre-unconsciousness manifestations of hepatic encephalopathy, and provide appropriate treatment.